Welcome to BIGA. Despite being in the early stages of development, our platform is equipped with a diverse range of tools and methods for genetic and epidemiological analysis. We recognize the dynamic and continuous development in these fields. Therefore, our team is committed to updating and expanding our platform to incorporate more advanced and useful tools. We are always interested in hearing back from you. If you have recommendations for additional tools or methods that could enrich our platform, please feel free to share them with us. Your insights and expertise will undoubtedly contribute to the development of our platform and benefit other users worldwide. This is an ongoing project for us, so stay tuned for more exciting updates and advancements in the future. We appreciate your patience and your support as we work diligently to implement more methods and improve your experience on our site.
BIGA GWAS Team
Documentation of BIGA GWAS
This guide is created as a reference for navigating our platform's diverse features with ease. In this manual, you will find an outline of the data specifications needed to get started, a walkthrough of the platform's backend configurations, and an explanation of all the functionalities.
Table of Contents
1. Data Processing
1.1 Data Processing Diagram1.2 Summary Statistics File Format and Content requirements
1.3 Build Input Files
1.4 Data Harmonization Pipeline
1.5 Harmonization Codes
1.6 Query Data from GWAS Catalog
1.7 Query Data from IEU OpenGWAS
1.8 Query Data from Neale Lab
2. Curated Datasets
2.1 Brain and Organ Imaging Traits2.2 UK Biobank (Neale Lab)
2.3 UKB Oxford Brain Imaging Traits
2.4 Biobank Japan
2.5 UKB-PPP European Protein GWAS
2.6 FinnGen
2.7 PGC (Psychiatric Genomics Consortium) and Other Brain Disorders
2.8 GWAS Catalog
2.9 CHIMGEN
3. Analysis
3.1 Massive LDSC Analysis3.2 Massive SumHer Analysis
3.3 Massive LAVA Analysis
3.4 Massive POPCORN Analysis
3.5 Pairwise Analysis
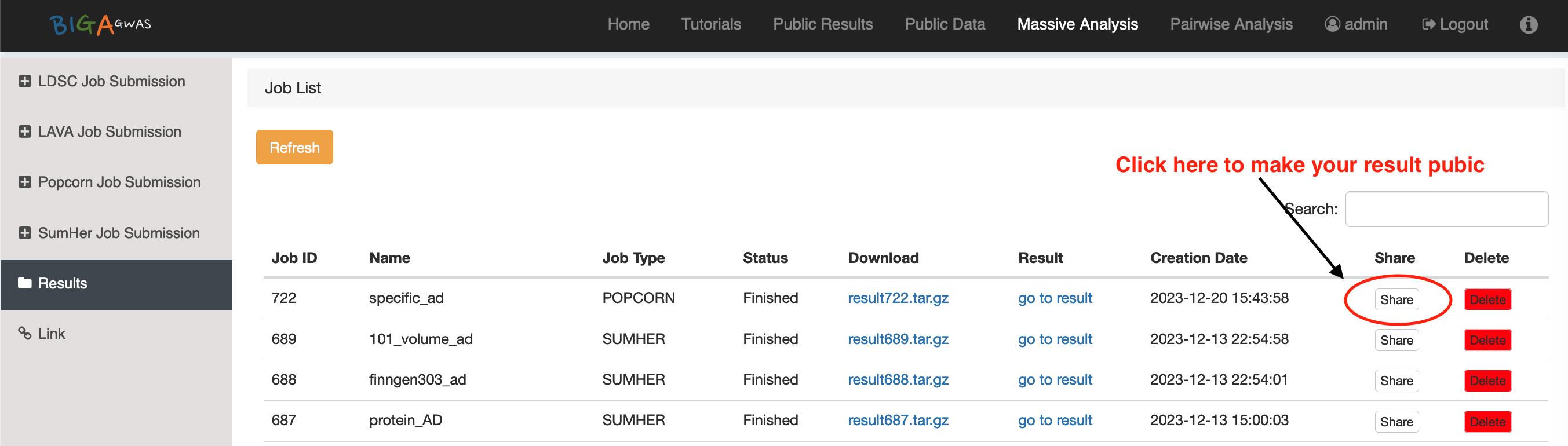
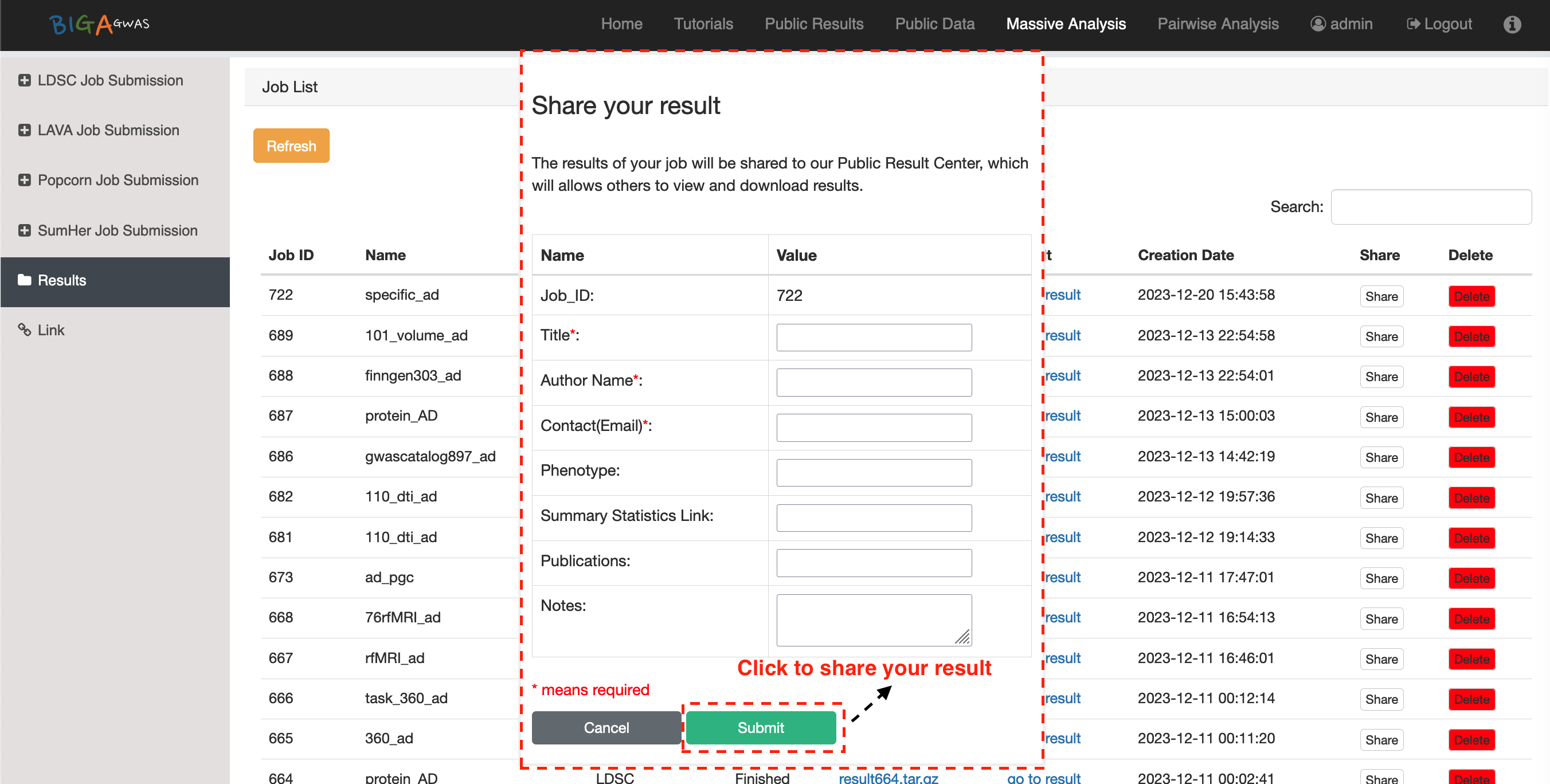
4. Analysis Results and Log Files
4.1 Analysis Results4.2 BIGA Log Files
5. Queue System and Periodic Jobs
5.1 Queue System5.2 Periodic Jobs
6. Other Functions
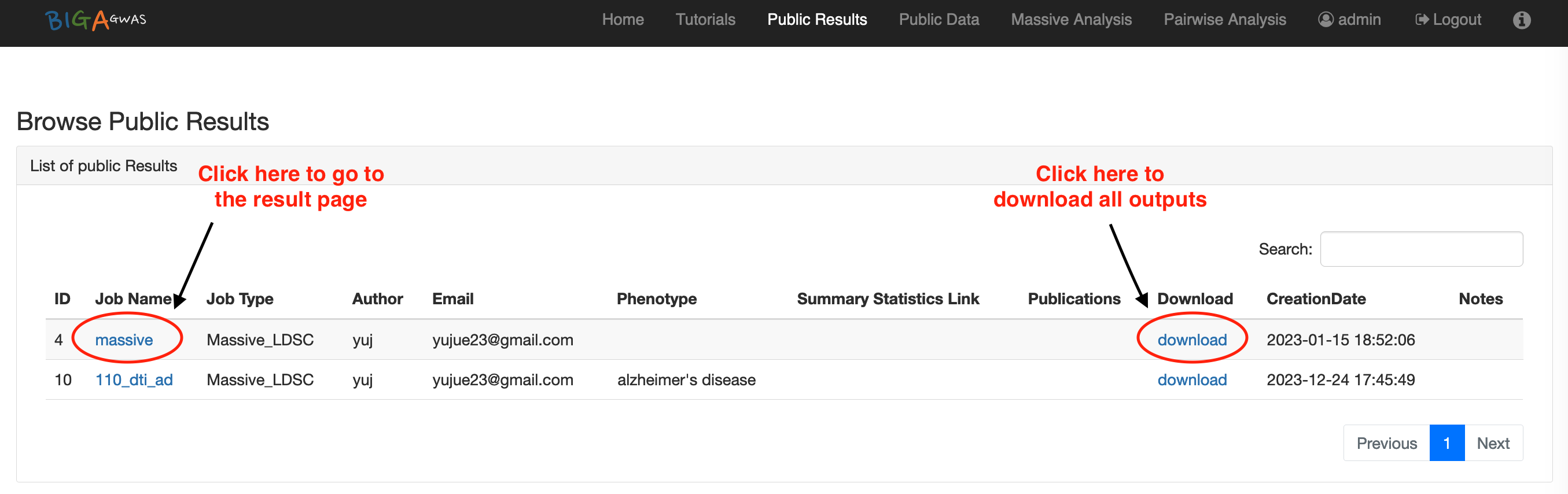
6.1 Email Notification6.2 Public Data
6.3 Public Results
7. Description examples
7.1 Method: LDSC7.2 Method: SumHer
7.3 Method: LAVA
7.4 Method: Popcorn
1. Data Processing
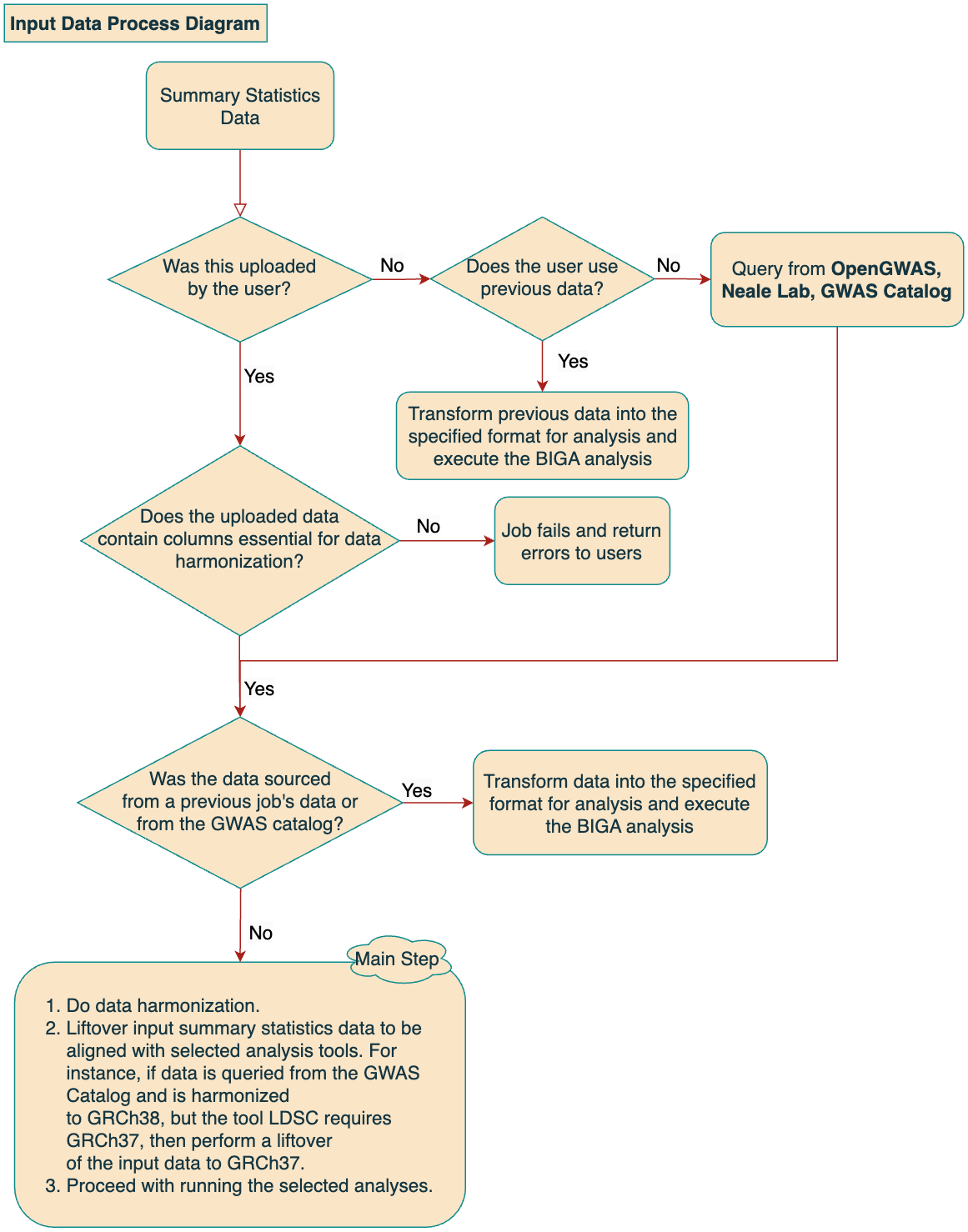
1.1 Data Processing Diagram
1.2 Table 1. Required and optional columns for harmonization and BIGA analysis
| Index | Column Name | Description | Type |
| 1 | chr | Indicates the chromosome where the single nucleotide polymorphism (SNP) is located | Require either chr,position or snp column. |
| 2 | position | Refers to the specific base pair position of the SNP on the chromosome | |
| 3 | snp | Identifier for the SNP (e.g.,rsID) | |
| 4 | a1 | Effect allele (allele associated with the risk of disease) | Mandatory |
| 5 | a2 | Other allele (non-effect allele) | Mandatory |
| 6 | beta | Refers to the estimated effect size of a particular genetic variant on a trait or disease. (beta > 0 means that effect allele (a1) increases disease risk). | Require either beta or odds_ratio column |
| 6 | odds_ratio | Odds ratio (1 indicates no effect; above 1 indicates effect allele (a1) increases disease risk) | |
| 7 | se | Standard error, a smaller standard error indicates a more reliable estimate of the beta/odds_ratio coefficient | Mandatory |
| 8 | eaf | Effect allele frequency, the frequency of the effect allele (a1) in the study population | Optional |
| 9 | pvalue | Indicates the statistical significance of the association between the SNP and the trait being studied | Mandatory |
| 10 | n | Sample size, the number of individuals or samples included in the study | Mandatory |
| 11 | z | Z-score (0 means no association between a genetic variant and disease risk; above than 0 indicates an increased risk associated with the variant) | Optional |
1.3 Build Input Files
1. Upload data
2. Use the Query data function
3. Use previous data
For the Upload data option, BIGA accepts files in .txt and .gz formats. For a gzipped file, please include only one plain text file. The uploaded file size should not exceed 600 Mb. The uploaded file can either be space-separated or tab-separated. Due to this format, please ensure that each element, including column names, does not contain any spaces. Additionally, make sure your data does not have repeated column names. Your input GWAS summary statistics should meet the column requirements, detailed in Table 1, to ensure proper data harmonization and further analysis. The headers of your uploaded file will be compared with the column requirements before harmonization. If the headers don't match our requirements, the job will be rejected. It is not necessary for the column names to be identical with the column names in Table 1. BIGA automatically identifies columns using similar to those used by the LDSC munge file. However, we recommend manually specifying as many column names as possible to prevent incorrect column matching.
Header Detection Rules
Column names are detected based on the headers provided. The headers to the left of the colon are interpreted as the header to the right of the colon (not case-sensitive). ). For example, “B”, “Beta”, “Effects”, or “Effect” will be interpreted as “beta”.• Chr | Chromosome :
chr• Position | Pos | Base_pair_location| BP :
position• Snp | Snpid | Snpid_UKB | RS | Rsid | Rs_id :
snp• P | Pvalue | P_value | Pval | P_val | GC_Pvalue :
pvalue• Effect_allele | A1 | allele1 | allele_1 | alt_allele | EA :
a1• A2 | Allele2 | Allele_2 | Ref_allele | Other_allele | NEA :
a2• NMISS | N :
n• Standard_error | SE :
se• OR | Odds_ratio :
odds_ratio• B | Beta | Effects | Effect :
beta• Zscore| Z-score | GC_Zscore | Z :
z• AF1 | Effect_allele_frequency | Eaf | FRQ | Maf | FRQ_U | F_U :
eafMissing value
Missing value can be written as "." or "NA" in the input file.Parameters
Specify the Human Genome Build version (GRCh37 or GRCh38) if your data does not include asnp
column for rsID and instead includes chromosome and position columns.For data built on GRCh37, BIGA will convert it to GRCh38 using the
liftOver Python package with the
chainfile provided by UCSC (https://hgdownload.cse.ucsc.edu/goldenpath/hg19/liftOver/, hg19ToHg38.over.chain.gz). Rows that cannot be aligned to a new position in GRCh38 will be removed.For data built on GRCh38, BIGA will skip chromosome and position alignment process with the reference chainfile.
The trait name of your uploaded file should be specified, please ensure your trait name does not include any spaces.
The population (European or East Asian) of your uploaded file should be specified.
Notes
BIGA assumes that theposition field in the uploaded file uses a 1-based coordinate system.
Rows containing chromosome X, Y, or M will also be excluded from the process. The pipeline currently
supports human genome version GRCh37 and GRCh38. If your data is created based on an older
version of the human genome e.g. NCBI36, you will need to update the genomic positions using the liftOver
tool from UCSC. However, if your data includes snp column, position update is not
required. BIGA will automatically update the chromosome and position information to align with the latest
dbSNP build 156 version.
In analyses like LDSC, LAVA, POPCORN, and SumHer, the effect_allele and
other_allele columns are interpreted as A1 and A2, respectively. If
both odds_ratio and beta are present, LDSC uses beta as the signed
summary statistics.
For Query data option, BIGA can directly query data from three external data resources:
1. GWAS Catalog
2. IEU OpenGWAS
3. Neale Lab
GWAS Catalog Data
BIGA supports querying of data from the GWAS Catalog, where the data should be FTP accessible and should already be harmonized by GWAS Catalog platform. BIGA provides a daily-updated GWAS Catalog harmonized list (Table 2). Since the data has been harmonized, BIGA will skip the harmonization step. You are required to input only the trait ID and sample size, after which BIGA will manage the FTP download. Once downloaded, BIGA converts the data into its universal format, incorporating columnssnp, chr,
position, pvalue, eaf, a1, a2,
n, odds_ratio or beta, and se. The harmonized file from
the GWAS Catalog might present diverse column names to represent harmonized columns. For instance, beta
could be labeled as hm_beta or beta. BIGA first checks for hm_beta;
if absent, it defaults to the beta column. Queries will be rejected if the proportion of
missing values in any harmonized column exceeds 50% or if essential fields are missing.
IEU OpenGWAS Data
BIGA supports querying of data from IEU OpenGWAS, where the data should be downloadable via the OpenGWAS website. You need to input the trait ID and the sample size, which can be found in OpenGWAS. The download files include a summary statistics VCF file and its corresponding index file. BIGA then transforms the original VCF files into its universal format, interpretingID as snp, CHROM as
chr, POS as position, LP as negative log p-value, AF1
as eaf, ALT as a1, REF as a2,
ES as beta (If the trait is binary, logOR will be interpreted as beta), and SE as se. Given that IEU OpenGWAS data might
not be harmonized, BIGA performs the harmonization step for any data acquired from this source. Since the
original file includes a snp column, specifying the human genome build version is unnecessary.
Additionally, data missing the required columns or with more than 80% missing values will not be
successfully queried.Note: If a user queries the trait from IEU OpenGWAS, the harmonized data will be stored on our cloud server. Should another user select this data, BIGA will use it directly instead of re-querying it.
Neale Lab Data
Data from Neale Lab has been pre-downloaded and harmonized by BIGA for your convenience. You can find a comprehensive list of available traits in Table 3, which includes both Trait IDs and Phenotype descriptions. To facilitate your search for a specific trait or disease, you can simply enter a trait, such asbmi, into the search field. This action will display all traits containing
bmi. Select the trait that suits your research needs. Given that this data is already
harmonized and stored locally within BIGA, querying data from Neale Lab is most time-saving among the
three external resources.For Previous data option, BIGA uses data from previous jobs as input files.
This function enables the use of previously processed data. In the 'Previous data' panel, you'll find a list of job IDs along with the trait names you've entered before. Utilizing this function bypasses the harmonization steps, as the data has already been harmonized. It will convert the data from the BIGA standard format into the format required by the specific method you're using. By doing so, it reduces the time spent on repetitive data requests and harmonization. Currently we keep user-input data for 7 days.
1.4 Data Harmonization Pipeline
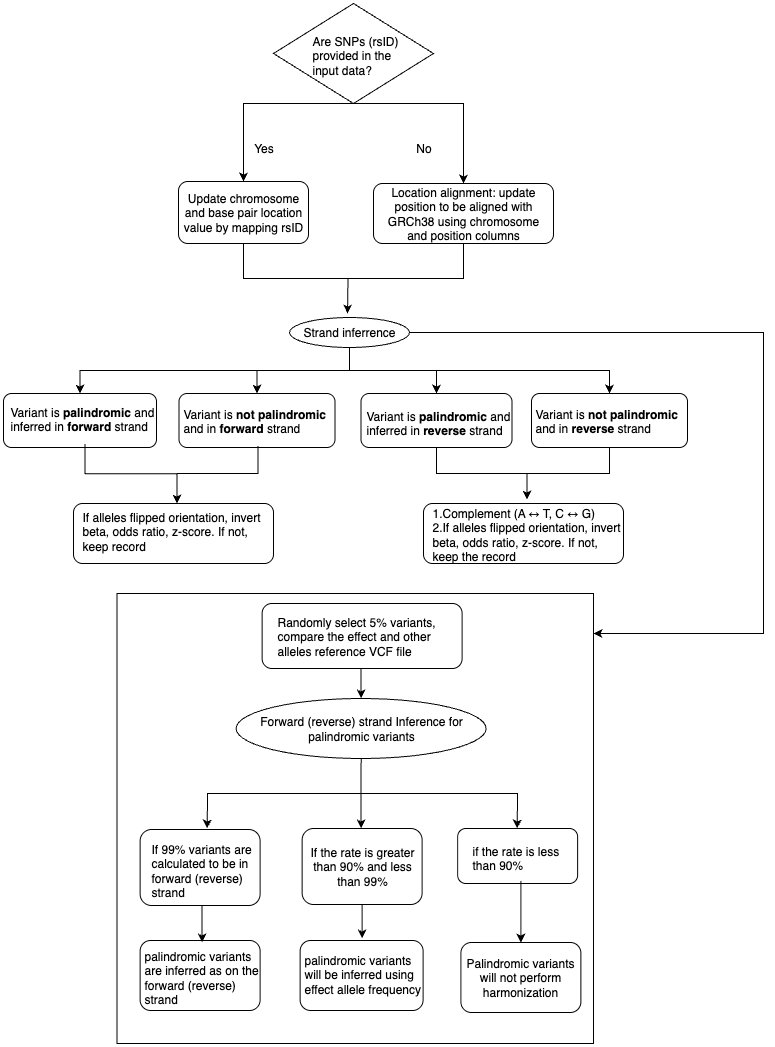
2. The main steps of harmonization includes:
(1) Update chromosome and base pair location value by mapping rsID if input data include snp (rsID) column.
(2) Location alignment: update position to be aligned with GRCh38 using chromosome and position columns if snp column is not provided. Remove variants if any of chromosome and position values are missing.
(3) Forward (reverse) strand Inference for palindromic variants:
a. Randomly select 5% variants, compare the effect and other alleles with corresponding alternative and reference alleles in the reference VCF file (LINK).
b. If 99% variants are calculated to be in forward (reverse) strand, then palindromic variants are inferred as on the forward (reverse) strand.
c. If the rate is greater than 90% and less than 99%, palindromic variants will be inferred using effect allele frequency (if not provided, palindromic variants will not perform harmonization).
d. Palindromic variants will not perform harmonization if the rate is less than 90%.
(4) Variant harmonization: using chromosome, position, effect and other alleles and effect allele frequency (eaf) (if provided) to be compared with ensemble VCF reference file.
a. Variant is palindromic and the strand needs to be inferred: if the eaf in varint and the eaf of alternative allele in reference VCF file are concordant(both greater or less than 0.5), the variant is in forward strand. Otherwise, the variant is in reverse strand.
b. If the palindromic variant is inferred in forward strand: if alleles flipped orientation, invert beta, odds ratio, z-score. If not, keep record.
c. If the palindromic variant is inferred in reverse strand: after complementing, if alleles flipped orientation, invert beta, odds ratio, z-score. If not, keep the record after complementing.
d. Variant is not palindromic and in forward strand: If alleles flipped orientation, invert beta, odds ratio, z-score. If not, keep record.
e. Variant is not palindromic and in reverse strand: after complementing, if alleles flipped orientation, invert beta, odds ratio, z-score. If not, keep the record after complementing.
f. Variants that do not match reference VCF file will be skipped.
(5) RsID: If input data does not include snp (rsID) column, update snp from chromosome, position, effect allele, other allele columns by referring the latest ensemble VCF reference file.
(6) QC: Invalid records are filtered out.
3. Liftover to GRCh37: After processing through our harmonization pipeline, the summary statistics data will be standardized to the GRCh38 build, regardless of their original genome build. However, considering that all analysis tools (such as LDSC and SumHer) work with data in GRCh37, we will use the liftover Python package with the chainfile provided by UCSC to convert position from GRCh38 to GRCh37 version.
1.5 Harmonization Codes
hm_code appended at the last column in your harmonized summary statistics.
Each row has a harmonization code. Please note that chromosome X, Y and MT are not included in our harmonized summary statistics.| Code | Description of Code |
| 1 | Palindromic; Infer strand; Forward strand; Alleles correct |
| 2 | Palindromic; Infer strand; Forward strand; Flipped alleles |
| 3 | Palindromic; Infer strand; Reverse strand; Alleles correct |
| 4 | Palindromic; Infer strand; Reverse strand; Flipped alleles |
| 5 | Palindromic; Assume forward strand; Alleles correct |
| 6 | Palindromic; Assume forward strand; Flipped alleles |
| 7 | Palindromic; Assume reverse strand; Alleles correct |
| 8 | Palindromic; Assume reverse strand; Flipped alleles |
| 9 | Palindromic; Drop palindromic; Not orientated |
| 10 | Forward strand; Alleles correct |
| 11 | Forward strand; Flipped alleles |
| 12 | Reverse strand; Alleles correct |
| 13 | Reverse strand; Flipped alleles |
| 14 | Required fields are not known; Not orientated |
| 15 | No matching variants in reference VCF; Not orientated |
| 16 | Multiple matching variants in reference VCF; Not orientated |
| 17 | Palindromic; Infer strand; EAF or reference VCF AF not known; Not orientated |
| 18 | Palindromic; Infer strand; EAF < specified minor allele frequency threshold; Not orientated |
Tips for interpreting terms used in harmonization:
1. Palindromic variant: A palindromic variant refers to a SNP where the effect allele and the other allele are complementary to each other on opposite DNA strands, making it ambiguous which allele is the effect allele without additional context. This happens because, in palindromic SNPs, the alleles can be represented as A/T or C/G pairs, where each allele looks the same when read forward or backward, just like a palindrome. This ambiguity arises in the context of GWAS summary statistics, especially when data from different studies or platforms are combined without specifying the reference strand.
2. Forward/reverse strand: In genetics, DNA is made of two complementary strands, known as the forward strand and the reverse strand, which run in opposite directions. The reference VCF file of dbSNP 156, the one BIGA use for harmonization, is in the forward strand orientation. BIGA map alleles in summary statistics to be aligned with alleles in this reference file. For example, the SNP with alleles A (effect allele) and G (other allele) is recorded in dbSNP 156 reference file. Study 1 reports the same SNP but lists the alleles as T (effect allele) and C (other allele), aligning with the reverse strand. We infer the SNP in study 1 is in reverse strand.
3. Flipped alleles: "flipped alleles" occur when the alleles of a genetic variant reported in a study are the reverse of those in the reference file, leading to inconsistencies. For example, the SNP with alleles A (effect allele) and G (other allele) is recorded in dbSNP 156 reference file. Study 1 reports the same SNP but lists the alleles as G (effect allele) and A (other allele). We infer the SNP in study 1 has flipped alleles.
1.6 Query Data from GWAS Catalog
To simplify and clarify the steps for querying summary statistics data from the GWAS Catalog, we can break down the process into three steps:
Step 1: Searching for the Trait
• Go to the GWAS Catalog's search panel.
• Enter the name or description of the phenotype or disease you're interested in.
• Navigate to the traits page and then find the trait you are interested in.
• The first instruction figure below gives an example of finding traits related to stroke.
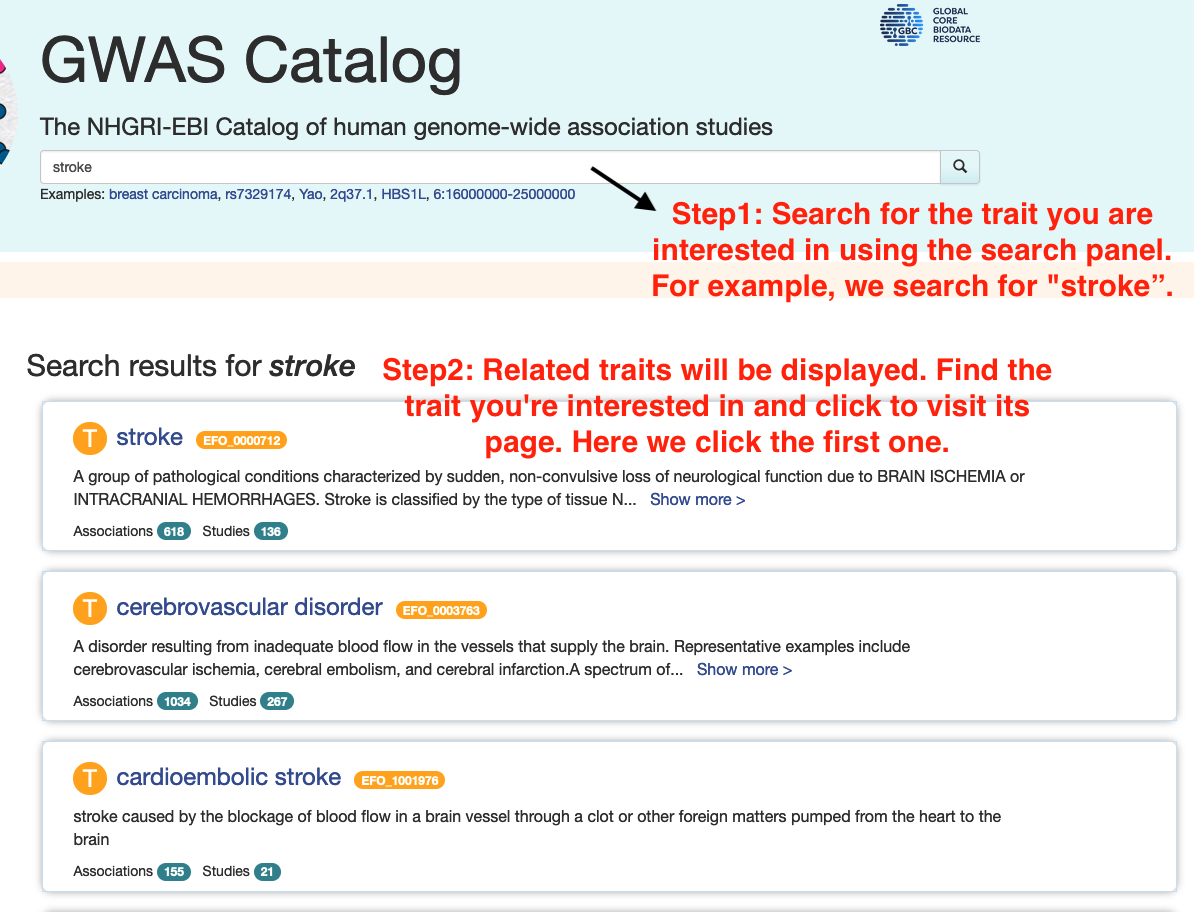

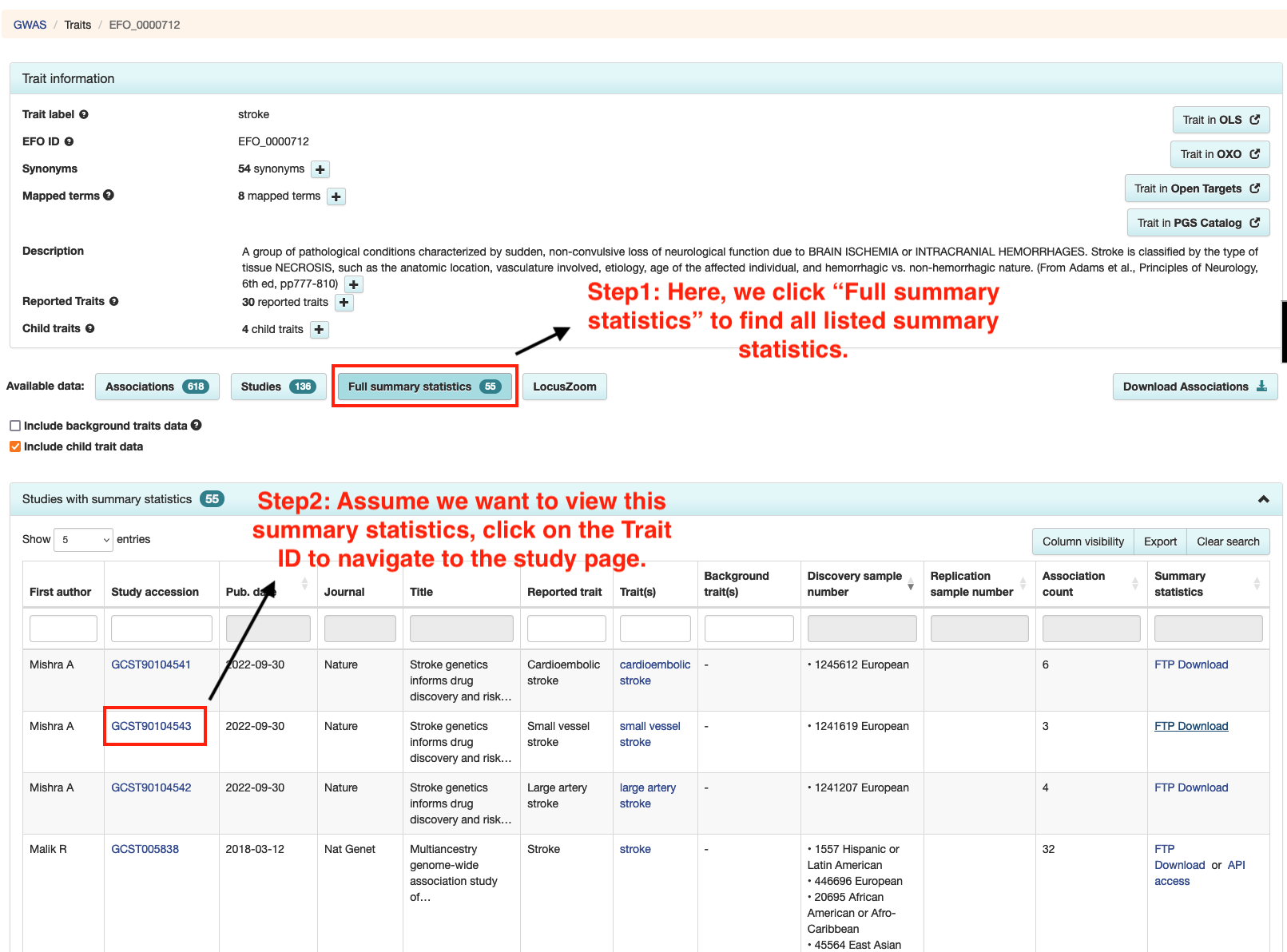
Step 2: Verifying Data Availability
• Navigate to the study page for your chosen trait, which will be labeled with "Study: [Trait ID]". This page will provide important details like sample size and population, which are necessary for your query.
• On the studies page, look for the "Full Summary Statistics" section.
• If there's an "FTP Download" link, this indicates that the original data is available.
• Click this link to check if the data has been harmonized. A harmonized dataset will have a "harmonised" folder on the FTP site, indicating it's ready for querying with BIGA.
• The second instruction figure below will help you identify the "FTP Download" link and the "harmonised" folder.
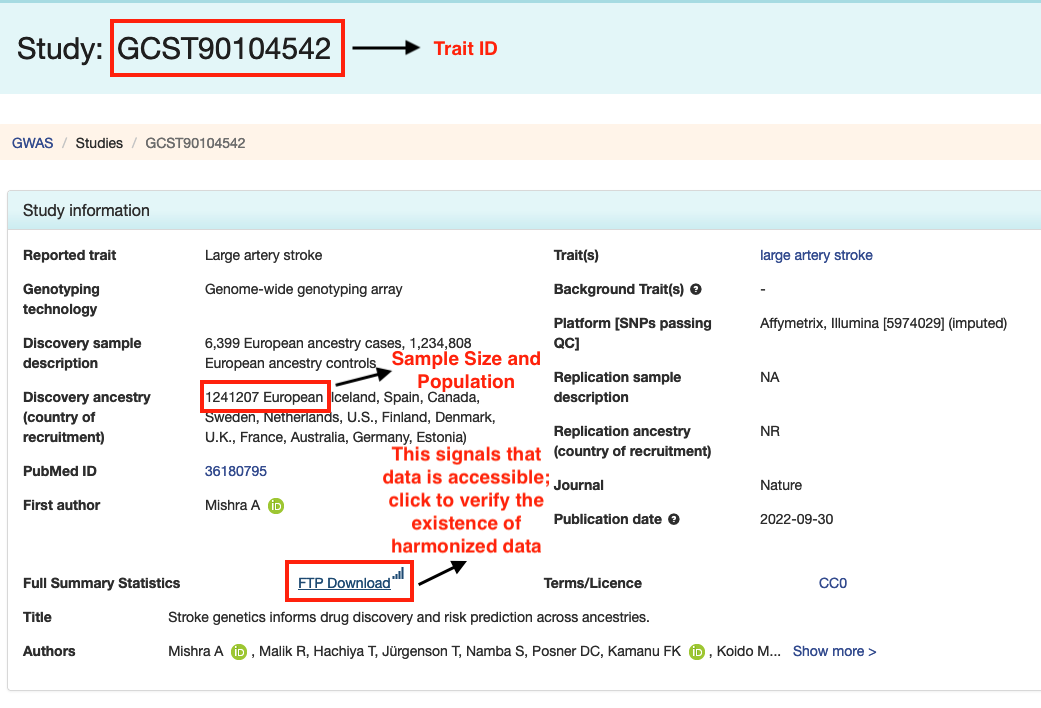

Step 3: Confirming the trait is query-able by BIGA
• In the table below, we list all available summary statistics that is query-able by BIGA.
• Search this table using the Trait ID from Step 1.
• If your trait is listed, it confirms that the data can be queried using BIGA.
Note: This table is updated daily at 5:30am EST.
1.7 Query Data from IEU OpenGWAS
To simplify and clarify the steps for querying summary statistics data from the IEU OpenGWAS, we can break down the process into two steps:
Step 1: Searching for the Trait
• Go to the IEU OpenGWAS search panel as shown in the first instruction figure below.
• At the section of Trait contain, enter the name or description of the phenotype or disease you're interested in.
• Navigate to the data page for your chosen trait, which will be labeled with "Phenotype Name and Dataset: [Trait ID]". This page will provide important details like sample size, population and human genome build version, which are necessary for your query.
• The data page is similar to the second instruction page below.
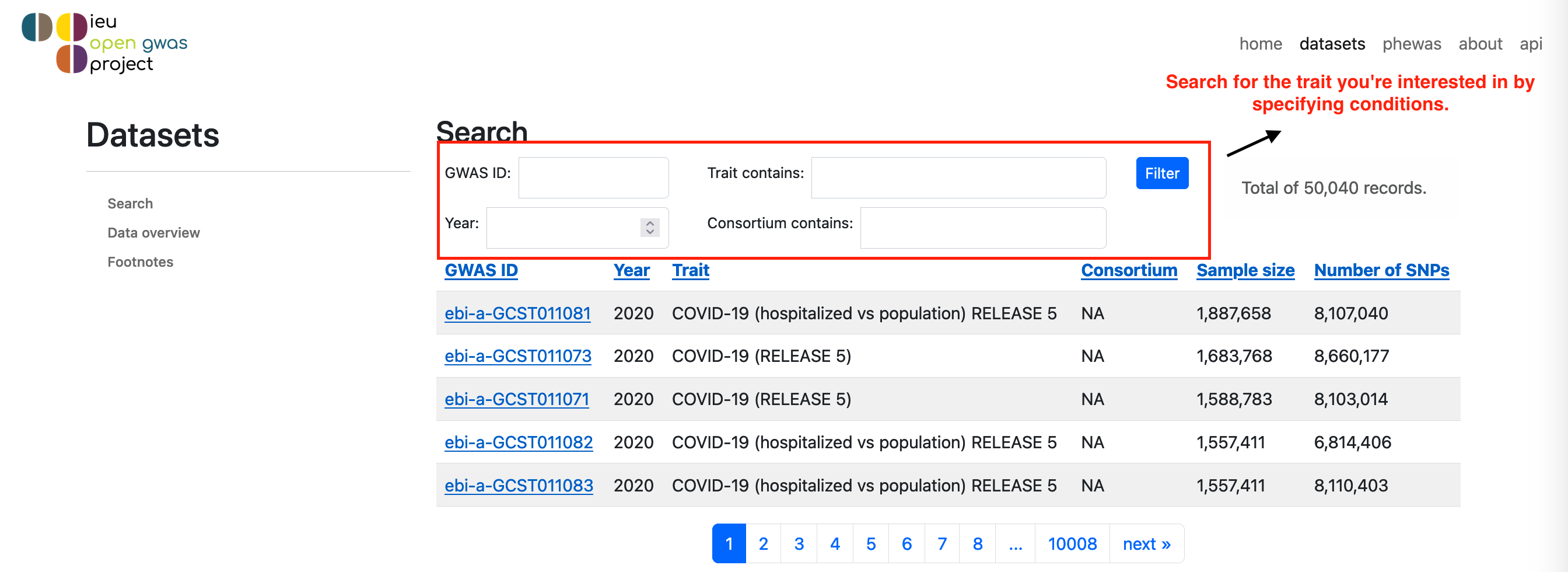

Step 2: Confirming the trait is query-able by BIGA
• If the "Download VCF" and "Download Index" blue buttons appear, the summary statistics data is query-able by BIGA.
• Or you can search trait ID or trait description directly using the table below, we list all available summary statistics that is query-able by BIGA.
• To proceed, enter the trait ID, population, and sample size into the query panel, and select the appropriate human genome build version.
Note: The table is updated monthly at 1:30 AM EST on the 11th day of each month.
1.8 Query Data from Neale Lab
The Neale Lab datasets have undergone data harmonization. More details about our data processing can be found at UK Biobank (Neale Lab).
The table below lists all the Neale Lab summary statistics data available for querying.
To find the data you are interested in, you can directly search by phenotype name or trait ID. In the panel of querying,
simply entering the trait ID is sufficient.
For more detailed information about the traits, please refer to Neale Lab's website.
2. Curated Dataset
2.1 Brain and Organ Imaging Traits
Description
BIG-KP is a knowledge portal dedicated to the study of various structural and functional imaging traits of the human brain and other organs. This dataset includes seven distinct sections:1. Regional brain volumes (n = 101)
2. Diffusion tensor imaging (DTI) parameters (n =110)
3. Resting-state fMRI (rfMRI) traits (n = 1080)
4. Task-related fMRI (tfMRI) traits (n = 919)
5. Regional rfMRI amplitude traits (n= 360)
6. Cardiac imaging traits (n =82)
Trait descriptions can be downloaded here.
Reference
For more information, please visit BIG-KP.Data Processing
Harmonized by BIGA, the original files have a header with columnsCHR, SNP, POS,
A1, A2, N, AF1, BETA, SE,
P where position is based on the GRCh37 version. These columns are reinterpreted in BIGA's
format as chr, snp, position, a1, a2,
n, eaf, beta, se, pvalue.
Since the snp column is provided, BIGA maps chromosomes and positions by referring to the latest
dbSNP build 156 version file when the snp is the same. Next, BIGA infers the
orientation (forward or reverse) of palindromic variants by randomly selecting 5% of variants for each
chromosome separately, as shown in the log file.
Chromosome 1: Forward strand rate: 0.998836, Reverse strand rate: 0.001164
Chromosome 2: Forward strand rate: 0.999861, Reverse strand rate: 0.000139
Chromosome 3: Forward strand rate: 0.999919, Reverse strand rate: 8.1e-05
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 0.996204, Reverse strand rate: 0.003796
Chromosome 7: Forward strand rate: 0.998482, Reverse strand rate: 0.001518
Chromosome 8: Forward strand rate: 0.999525, Reverse strand rate: 0.000475
Chromosome 9: Forward strand rate: 0.998611, Reverse strand rate: 0.001389
Chromosome 10: Forward strand rate: 0.994752, Reverse strand rate: 0.005248
Chromosome 11: Forward strand rate: 0.994062, Reverse strand rate: 0.005938
Chromosome 12: Forward strand rate: 0.999817, Reverse strand rate: 0.000183
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 0.993571, Reverse strand rate: 0.006429
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 0.99989, Reverse strand rate: 0.00011
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 22: Forward strand rate: 1.0, Reverse strand rate: 0.0
Overall Forward strand rate: 0.998702, Reverse strand rate:0.001298
The script above is the log file of ukb_phase1to3_roi_volume101_june_2020_pheno_9 trait (one
trait from the Regional brain volumes group), it's observed that the forward strand rate is above 99%. Therefore,
palindromic variants are inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 380854, 'Forward strand; Flipped orientation; Requires harmonisation': 86197, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 67744, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 15806, 'Reverse strand; Correct orientation; Already harmonised': 1193, 'Reverse strand; Flipped orientation; Requires harmonisation': 234})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 380854. It indicates that 380854 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Forward strand; Flipped orientation; Requires harmonisation: 86197. This indicates 86199 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds
ratio, z-score if provided.Palindromic; Assume forward strand; Correct orientation; Already harmonised: 67744. This means it's assumed they are on the forward strand and are in the correct orientation, so no further harmonization is needed.
Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 15806. This means it's assumed they are on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if provided.Reverse strand; Correct orientation; Already harmonised': 1193 This means that 1193 SNPs are on reverse strand but are in the correct orientation as per the reference. They are already harmonized, so BIGA will keep the records.
Reverse strand; Flipped orientation; Requires harmonisation: 234.These 234 SNPs are on the reverse strand and have a flipped orientation, which require harmonization. BIGA will invert
beta,
odds ratio, z-score if provided.The harmonization of each chromosome is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column. To
accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer for a human genome version of
GRCh37, BIGA converts the position data from GRCh38 to GRCh37. Then,
we get the unified format of input file in BIGA.
2.2 UK Biobank (Neale Lab)
Description
The Neale Lab dataset includes imputed genotypes from HRC plus UK10K & 1000 Genomes reference panels released by UK Biobank in March 2018. This dataset includes 4347 phenotypes categorized under the 'both_sexes' classification and excludes traits marked with an 'pending' status in md5s.Phenotype description can be downloaded here.
Reference
For more information, please visit Neale Lab.Data Processing
Harmonized by BIGA, the original files have a header with columnsCHR, BP,
A1, A2, N, EAF, BETA, SE,
P where position is based on the GRCh37 version. These columns are reinterpreted in BIGA's
format as chr,position, a1, a2, n, eaf,
beta, se, pvalue. Without a snp column provided, the
snp column is retrieved during the harmonization step. BIGA maps GRCh37 to GRCh38
locations using a liftover chainfile from UCSC, as the reference file dbSNP build 156 version is built on
GRCh38 version. Next, BIGA infers the orientation (forward or reverse) of palindromic variants
by randomly selecting 5% of variants for each chromosome separately, as shown in the log file.
Chromosome 1: Forward strand rate: 0.999728, Reverse strand rate: 0.000272
Chromosome 2: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 3: Forward strand rate: 0.999973, Reverse strand rate: 2.7e-05
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 0.999323, Reverse strand rate: 0.000677
Chromosome 7: Forward strand rate: 0.9998, Reverse strand rate: 0.0002
Chromosome 8: Forward strand rate: 0.999858, Reverse strand rate: 0.000142
Chromosome 9: Forward strand rate: 0.999733, Reverse strand rate: 0.000267
Chromosome 10: Forward strand rate: 0.999042, Reverse strand rate: 0.000958
Chromosome 11: Forward strand rate: 0.999109, Reverse strand rate: 0.000891
Chromosome 12: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 0.999096, Reverse strand rate: 0.000904
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 0.99986, Reverse strand rate: 0.00014
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 22: Forward strand rate: 1.0, Reverse strand rate: 0.0
Overall Forward strand rate: 0.999778, Reverse strand rate:0.000222
The script above is the log file of Z47.gwas.imputed_v3.both_sexes trait (one trait from the
Neale Lab group), it can be observed that the forward strand rate is above 99%. Therefore, palindromic variants
are inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 684193, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 121370, 'Forward strand; Flipped orientation; Requires harmonisation': 1043, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 216, 'Reverse strand; Correct orientation; Already harmonised': 174, 'Reverse strand; Flipped orientation; Requires harmonisation': 143})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 684193. It indicates that 684193 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Palindromic; Assume forward strand; Correct orientation; Already harmonised: 121370. This means that they are assumed to be on the forward strand and are in the correct orientation, so no further harmonization is needed.
Forward strand; Flipped orientation; Requires harmonisation: 1043. This indicates 1043 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds
ratio, z-score if provided.Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 216. 216 SNPs fall into this category. This means that they are assumed to be on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if
provided.Reverse strand; Correct orientation; Already harmonised': 174 This means that 174 SNPs are on reverse strand but are in the correct orientation as per the reference. They are already harmonized. BIGA will keep the records.
Reverse strand; Flipped orientation; Requires harmonisation: 143.These 143 SNPs are on the reverse strand and have a flipped orientation, which require harmonization. BIGA will invert
beta,
odds ratio, z-score if provided.Each chromosome harmonization is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column. To
accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version of
GRCh37, BIGA converts the position data from GRCh38 to GRCh37. Then,
we get the unified format of input file in BIGA.2.3 UKB Oxford Brain Imaging Traits
Description
The UKB Oxford Brain Imaging Traits is a collection of neuroimaging data traits derived from the UK Biobank. This dataset includes five distinct sections:1. Structural MRI (SMRI, n=1437)
2. Difussion MRI (dMRI, n=675)
3. Task functional MRI (tfMRI, n=16)
4. Resting functional MRI (rfMRI, amplitude, n=76)
5. Resting functional MRI (rfMRI, functional connectivity, n=1701)
Trait descriptions can be downloaded here.
Reference
For more information, please visit UKB Oxford.Data Processing
Harmonized by BIGA, the original files have a header with columnsCHR,SNP, POS,
A1, A2, N, AF1, BETA, SE,
P where position is based on the GRCh37 version. These columns are reinterpreted in BIGA's
format as chr, snp, position, a1, a2,
n, eaf, beta, se, pvalue. With the snp
column provided, BIGA maps snp to chromosome and position by referencing the latest reference
file. Next, BIGA infers the orientation (forward or reverse) of palindromic variants by randomly selecting 5%
of variants for each chromosome separately, as shown in the log file.
Chromosome 1: Forward strand rate: 0.998876, Reverse strand rate: 0.001124
Chromosome 2: Forward strand rate: 0.999965, Reverse strand rate: 3.5e-05
Chromosome 3: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 0.996888, Reverse strand rate: 0.003112
Chromosome 7: Forward strand rate: 0.99783, Reverse strand rate: 0.00217
Chromosome 8: Forward strand rate: 0.99926, Reverse strand rate: 0.00074
Chromosome 9: Forward strand rate: 0.998916, Reverse strand rate: 0.001084
Chromosome 10: Forward strand rate: 0.995177, Reverse strand rate: 0.004823
Chromosome 11: Forward strand rate: 0.994628, Reverse strand rate: 0.005372
Chromosome 12: Forward strand rate: 0.999821, Reverse strand rate: 0.000179
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 0.994196, Reverse strand rate: 0.005804
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 22: Forward strand rate: 0.999575, Reverse strand rate: 0.000425
Overall Forward strand rate: 0.998788, Reverse strand rate:0.001212
The script above is the log file of ukb_smith_dMRI_2_imputed_pheno9 trait (one trait from the
dMRI group of UKB Oxford), it's observed that the forward strand rate is above 99%. Therefore, palindromic
variants are inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 380854, 'Forward strand; Flipped orientation; Requires harmonisation': 86199, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 67743, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 15807, 'Reverse strand; Correct orientation; Already harmonised': 1193, 'Reverse strand; Flipped orientation; Requires harmonisation': 234})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 380854. It indicates that 380854 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Forward strand; Flipped orientation; Requires harmonisation: 86199 This means 86199 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds
ratio, z-score if provided.Palindromic; Assume forward strand; Correct orientation; Already harmonised: 67743. This means it's assumed they are on the forward strand and are in the correct orientation, so no further harmonization is needed.
Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 15807. This means it's assumed they are on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if provided.Reverse strand; Correct orientation; Already harmonised': 1193 This means that 1193 SNPs are on reverse strand but are in the correct orientation as per the reference. They are already harmonized. BIGA will keep the records.
Reverse strand; Flipped orientation; Requires harmonisation: 234.These 234 SNPs are on the reverse strand and have a flipped orientation, which require harmonization. BIGA will invert
beta,
odds ratio, z-score if provided.Each chromosome harmonization is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column.
To accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version
of GRCh37, BIGA converts the position data from GRCh38 to GRCh37.
Then, we get the unified format of input file in BIGA.
2.4 Biobank Japan
Description
The Biobank Japan project is a large-scale biobank that collects genetic, clinical, and lifestyle information from a cohort of Japanese individuals. This dataset includes 177 phenotypes.Phenotype descriptions can be downloaded here.
Reference
For more information, please visit Biobank Japan.Data Processing
Harmonized by BIGA, the original files have a header with columnsCHR,SNPID, POS,
Allele1, Allele2, N, AF_Allele2, BETA,
SE, p.value where position is based on the GRCh37 version. Please note that Allele2
is the effect allele. These columns are reinterpreted in BIGA's format as chr, snp,
position, a2, a1, n, eaf, beta,
se, pvalue. With the snp column provided, BIGA maps snp
to chromosome and position by referencing the latest reference file. Next, BIGA infers the
orientation (forward or reverse) of palindromic variants by randomly selecting 5% of variants for each
chromosome separately, as shown in the log file.
Chromosome 1: Forward strand rate: 0.999125, Reverse strand rate: 0.000875
Chromosome 2: Forward strand rate: 0.999894, Reverse strand rate: 0.000106
Chromosome 3: Forward strand rate: 0.99997, Reverse strand rate: 3e-05
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 0.997778, Reverse strand rate: 0.002222
Chromosome 7: Forward strand rate: 0.998396, Reverse strand rate: 0.001604
Chromosome 8: Forward strand rate: 0.999838, Reverse strand rate: 0.000162
Chromosome 9: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 10: Forward strand rate: 0.997203, Reverse strand rate: 0.002797
Chromosome 11: Forward strand rate: 0.998023, Reverse strand rate: 0.001977
Chromosome 12: Forward strand rate: 0.999909, Reverse strand rate: 9.1e-05
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 0.99764, Reverse strand rate: 0.00236
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 22: Forward strand rate: 0.999809, Reverse strand rate: 0.000191
Overall Forward strand rate: 0.999342, Reverse strand rate:0.000658
The script above is the log file of GWASsummary_Zoster_Japanese_SakaueKanai2020 trait (one
trait from the Biobank Japan), it's observed that the forward strand rate is above 99%. Therefore,
palindromic variants are inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 628517, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 103469, 'Reverse strand; Correct orientation; Already harmonised': 1256, 'Forward strand; Flipped orientation; Requires harmonisation': 779, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 356, 'Reverse strand; Flipped orientation; Requires harmonisation': 25})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 628517. It indicates that 628517 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Palindromic; Assume forward strand; Correct orientation; Already harmonised: 103469. This means it's assumed they are on the forward strand and are in the correct orientation, so no further harmonization is needed.
Reverse strand; Correct orientation; Already harmonised': 1256 This means that 1256 SNPs are on reverse strand but are in the correct orientation as per the reference. They are already harmonized. BIGA will keep the records.
Forward strand; Flipped orientation; Requires harmonisation: 779 This means 779 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds ratio,
z-score if provided.Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 356. This means it's assumed they are on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if provided.Reverse strand; Flipped orientation; Requires harmonisation: 25. These 25 SNPs are on the reverse strand and have a flipped orientation, which require harmonization. BIGA will invert
beta,
odds ratio, z-score if provided.Each chromosome harmonization is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column.
To accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version
of GRCh37, BIGA converts the position data from GRCh38 to GRCh37.
Then, we get the unified format of input file in BIGA.
2.5 UKB-PPP European Protein GWAS
Description
The UKB-PPP European Protein GWAS dataset refers to a collection of GWAS summary statistics of the protein expressions within a European population. This dataset includes 2940 proteins.Trait descriptions can be downloaded here.
Reference
For more information, please visit UKB-PPP.Data Processing
Harmonized by BIGA, the original files have a header with columnsCHROM,POS, ALLELE0,
ALLELE1, N, A1FREQ, BETA, SE,
P where position is based on the GRCh37 version. Please note that ALLELE1 is the
effect allele. These columns are reinterpreted in BIGA's format as chr, position,
a2, a1, n, eaf, beta, se,
pvalue. Without a snp column provided, the snp column is retrieved
during the harmonization step. BIGA maps hg19 to hg38 locations using a liftover chainfile from UCSC, as the
reference file dbSNP build 156 version is built on GRCh38 version. Next, BIGA infers the
orientation (forward or reverse) of palindromic variants by randomly selecting 5% of variants for each
chromosome separately, as shown in the log file.
Chromosome 1: Forward strand rate: 0.999762, Reverse strand rate: 0.000238
Chromosome 2: Forward strand rate: 0.99998, Reverse strand rate: 2e-05
Chromosome 3: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 0.999208, Reverse strand rate: 0.000792
Chromosome 7: Forward strand rate: 0.999884, Reverse strand rate: 0.000116
Chromosome 8: Forward strand rate: 0.999969, Reverse strand rate: 3.1e-05
Chromosome 9: Forward strand rate: 0.999528, Reverse strand rate: 0.000472
Chromosome 10: Forward strand rate: 0.998169, Reverse strand rate: 0.001831
Chromosome 11: Forward strand rate: 0.999345, Reverse strand rate: 0.000655
Chromosome 12: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 0.999345, Reverse strand rate: 0.000655
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 22: Forward strand rate: 1.0, Reverse strand rate: 0.0
Overall Forward strand rate: 0.999754, Reverse strand rate:0.000246
The script above is the log file of ZPR1_O75312_OID30801_v1_Neurology_II trait (one trait from
the UKB-PPP), it is observed that the forward strand rate is above 99%. Therefore, palindromic variants are
inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 784340, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 139553, 'Forward strand; Flipped orientation; Requires harmonisation': 932, 'Reverse strand; Correct orientation; Already harmonised': 205, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 187, 'Reverse strand; Flipped orientation; Requires harmonisation': 178})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 784340. It indicates that 784340 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Palindromic; Assume forward strand; Correct orientation; Already harmonised: 139553. This means it's assumed they are on the forward strand and are in the correct orientation, so no further harmonization is needed.
Forward strand; Flipped orientation; Requires harmonisation: 932 This means 932 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds ratio,
z-score if provided.Reverse strand; Correct orientation; Already harmonised: 205. A small number of SNPs (205) are on the reverse DNA strand but are in the correct orientation as per the reference. They are already harmonized. BIGA will keep the records.
Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 187. This means it's assumed they are on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if provided.Reverse strand; Flipped orientation; Requires harmonisation: 178. 178 SNPs fall into this category, where they are on the reverse strand and have a flipped orientation. These require harmonization. BIGA will invert
beta, odds ratio, z-score if provided.Each chromosome harmonization is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column.
To accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version
of GRCh37, BIGA converts the position data from GRCh38 to GRCh37.
Then, we get the unified format of input file in BIGA.
2.6 FinnGen
Description
The FinnGen dataset is a large-scale genetic research project combining Finnish biobank data with comprehensive health records. This dataset includes 303 selected diseases with the number of case larger than 10,000.Disease descriptions can be downloaded here.
Reference
For more information, please visit FinnGen.Data Processing
Harmonized by BIGA, the original files have a header with columnsCHR,SNP, POS,
effect_allele, other_allele, N, effect_allele_freqency,
beta, se, P where position is based on the GRCh37 version. These
columns are reinterpreted in BIGA's format as chr, snp, position,
a1, a2, n, eaf, beta, se,
pvalue. With the snp column provided, BIGA maps snp to chromosome and
position by referencing the latest reference file. Next, BIGA infers the orientation (forward or reverse) of
palindromic variants by randomly selecting 5% of variants for each chromosome separately, as shown in the
log file.
Chromosome 1: Forward strand rate: 0.999709, Reverse strand rate: 0.000291
Chromosome 2: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 3: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 7: Forward strand rate: 0.999907, Reverse strand rate: 9.3e-05
Chromosome 8: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 9: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 10: Forward strand rate: 0.999918, Reverse strand rate: 8.2e-05
Chromosome 11: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 12: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 0.999802, Reverse strand rate: 0.000198
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 22: Forward strand rate: 0.999707, Reverse strand rate: 0.000293
Overall Forward strand rate: 0.999958, Reverse strand rate:4.2e-05
The script above is the log file of finngen_R9_Z21_PROCREATIVE_MANAG_GRCh37_CrossMap trait (one
trait from the FinnGen), it's observed that the forward strand rate is above 99%. Therefore, palindromic
variants are inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 945824, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 166105, 'Forward strand; Flipped orientation; Requires harmonisation': 45, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 6, 'Reverse strand; Correct orientation; Already harmonised': 5})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 945824. It indicates that 945824 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Palindromic; Assume forward strand; Correct orientation; Already harmonised: 166105. This means it's assumed they are on the forward strand and are in the correct orientation, so no further harmonization is needed.
Forward strand; Flipped orientation; Requires harmonisation: 45 This means 45 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds ratio,
z-score if provided.Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 6. This means it's assumed they are on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if provided.Reverse strand; Correct orientation; Already harmonised: 5. A small number of SNPs (306) are on the reverse DNA strand but are in the correct orientation as per the reference. They are already harmonized. BIGA will keep the records.
Each chromosome harmonization is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column.
To accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version
of GRCh37, BIGA converts the position data from GRCh38 to GRCh37.
Then, we get the unified format of input file in BIGA.
2.7 PGC (Psychiatric Genomics Consortium) and Other Brain Disorders
Description
The PGC dataset represents a comprehensive collection of genetic and clinical data focused on psychiatric disorders. In addition, we also included a few brain disorders such as Alzheimer's disease, Stroke and its subtypes, and Depression. This dataset currently contains 24 phenotypes.Phenotype descriptions can be downloaded here.
Reference
For more information about PGC, please visit the PGC website.Data Processing
Partially harmonized by BIGA due to missing specific columns in some traits. Harmonized traits include ADHD, several bipolar disorder indices (pgc_bip2021_al, pgc_bip2021_BDI, pgc_bip2021_BDII), depression-related traits (depression_pgc_ukb, pts_eur_freeze2_overall), hoarding2022, and a series of traits related to schizophrenia (PGC3_SCZ_wave3, depression, GCST90104539-43, GCST90027158). Traits that remain unharmonized are TS_Oct2018, several opioid exposure comparisons within European ancestry cohorts, substance use disorder indices (CUD_EUR_full_public_11.14.2020, pgc_alcdep.eur_discovery.aug2018_release), ocd_aug2017, autism spectrum disorder traits (iPSYCH-PGC_ASD_Nov2017), and pgcAN2_2019_07.2.8 GWAS Catalog
Description
The GWAS Catalog is a comprehensive resource that provides access to a curated collection of GWAS results. This dataset includes 897 traits, curated to include those with substantial sample sizes (the number of case larger than 5,000 for diseases and the sample size larger than 10,000 for traits) . Datasets with high missing rate in beta, odds_ratio, se columns are removed.Phenotype descriptions can be downloaded here.
Reference
For more information, please visit GWAS Catalog.Data Processing
These data have been harmonized by GWAS Catalog. BIGA converts the pre-downloaded data into the universal format, incorporating columnssnp, chr, position, pvalue, eaf,
a1, a2, n, odds_ratio or beta, and
se. The harmonized file from the GWAS Catalog might present diverse column names to represent
harmonized columns. For instance, beta could be labeled as `hm_beta` or `beta`. BIGA first
checks for hm_beta; if absent, it defaults to the beta column.To accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version of
GRCh37, BIGA converts the position from GRCh38 to GRCh37. Then, we
get the unified format of input file in BIGA.
2.9 CHIMGEN
Description
The Chinese Imaging Genetics (CHIMGEN) study is a comprehensive neuroimaging genetics cohort in the Chinese population. This dataset includes 44 hippocampal volumetric traits.Trait descriptions can be downloaded here.
Reference
For more information, please visit CHIMGEN.Data Processing
Harmonized by BIGA, the original files have a header with columnschr,pos, Other_allele, Effect_allele, af_Effect_allele, beta, se, -log10P, P, N
where position is based on the GRCh37 version. pvalue is calculated from -log10P. These columns are reinterpreted in BIGA's format as chr, position, a2, a1, eaf, beta, se, pvalue, n. Without a snp column provided, the snp column is retrieved during the harmonization step. BIGA maps hg19 to hg38 locations using a liftover chainfile from UCSC, as the reference file dbSNP build 156 version is built on GRCh38 version. Next, BIGA infers the orientation (forward or reverse) of palindromic variants by randomly selecting 5% of variants for each chromosome separately, as shown in the log file.
Chromosome 1: Forward strand rate: 0.999758, Reverse strand rate: 0.000242
Chromosome 2: Forward strand rate: 0.99998, Reverse strand rate: 2e-05
Chromosome 3: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 4: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 5: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 6: Forward strand rate: 0.999614, Reverse strand rate: 0.000386
Chromosome 7: Forward strand rate: 0.999405, Reverse strand rate: 0.000595
Chromosome 8: Forward strand rate: 0.999934, Reverse strand rate: 6.6e-05
Chromosome 9: Forward strand rate: 0.999876, Reverse strand rate: 0.000124
Chromosome 10: Forward strand rate: 0.998454, Reverse strand rate: 0.001546
Chromosome 11: Forward strand rate: 0.99927, Reverse strand rate: 0.00073
Chromosome 12: Forward strand rate: 0.999929, Reverse strand rate: 7.1e-05
Chromosome 13: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 14: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 15: Forward strand rate: 0.998746, Reverse strand rate: 0.001254
Chromosome 16: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 17: Forward strand rate: 0.999937, Reverse strand rate: 6.3e-05
Chromosome 18: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 19: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 20: Forward strand rate: 1.0, Reverse strand rate: 0.0
Chromosome 21: Forward strand rate: 0.999867, Reverse strand rate: 0.000133
Chromosome 22: Forward strand rate: 0.999744, Reverse strand rate: 0.000256
Overall Forward strand rate: 0.999748, Reverse strand rate:0.000252
The script above is the log file of R_subiculum_head trait (one
trait from the CHIMGEN), it's observed that the forward strand rate is above 99%. Therefore, palindromic
variants are inferred to be on the forward strand.
Chromosome 6, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 775133, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 144108, 'Forward strand; Flipped orientation; Requires harmonisation': 1035, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 216, 'Reverse strand; Correct orientation; Already harmonised': 197, 'Reverse strand; Flipped orientation; Requires harmonisation': 171})
The above is the log file that summarizes the result of chromosome 6 during the harmonization.Forward strand; Correct orientation; Already harmonised: 775133. It indicates that 775133 SNPs are on the forward DNA strand and are already in the correct orientation. They do not require any further harmonization.
Palindromic; Assume forward strand; Correct orientation; Already harmonised: 144108. This means it's assumed they are on the forward strand and are in the correct orientation, so no further harmonization is needed.
Forward strand; Flipped orientation; Requires harmonisation: 1035 This means 1035 SNPs on the forward strand in a flipped orientation. Therefore, BIGA will invert
beta, odds ratio,
z-score if provided.Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation: 216. This means it's assumed they are on the forward strand but are in the flipped orientation. BIGA will invert
beta, odds ratio, z-score if provided.Reverse strand; Correct orientation; Already harmonised: 197. A small number of SNPs (197) are on the reverse DNA strand but are in the correct orientation as per the reference. They are already harmonized. BIGA will keep the records.
Reverse strand; Flipped orientation; Requires harmonisation: 171. These 171 SNPs are on the reverse strand and have a flipped orientation, which require harmonization. BIGA will invert
beta,
odds ratio, z-score if provided.Each chromosome harmonization is performed in parallel to save time. After harmonization, BIGA combines each chromosome and sorts harmonized data based on
chr and position column.
To accommodate the requirements of LDSC, LAVA, Popcorn, and SumHer, which all require a human genome version
of GRCh37, BIGA converts the position data from GRCh38 to GRCh37.
Then, we get the unified format of input file in BIGA.
3. Analysis Methods
3.1 Massive LDSC Analysis
Reference
LDSC: version 1.0.1Curated Dataset
BIGA offers 7 European datasets and 2 East Asian datasets for linkage disequilibrium score regression (LDSC) genetic correlation analysis. These datasets include:1. Brain and Organ Imaging Traits
2. PGC and and Other Brain Disorders
3. FinnGen
4. UK Biobank (Neale Lab)
5. Biobank Japan
6. UKB-Oxford Brain Imaging Traits
7. GWAS Catalog
8. UKB-PPP European Protein GWAS
9. CHIMGEN
Input File Build
The standard input file format in LDSC is .sumstats.gz. This format is achieved usingmunge_sumstats.py
from the LDSC software. The script parameters differ based on columns provided in the input file:
If both
beta and odds_ratio are present, BIGA prioritizes beta. The
followings are the BIGA's default running script:python2 $ldsc_path/munge_sumstats.py --sumstats $summary_statistics1 --out $output --merge-alleles $ldsc_path/w_hm3.snplist --chunksize 100000 --ignore 'odds_ratio'
python2 $ldsc_path/munge_sumstats.py --sumstats $summary_statistics1 --out $output --merge-alleles $ldsc_path/w_hm3.snplist --chunksize 100000
BIGA's format does not require an INFO column. LDSC used the HapMap3 variants (with the --merge-alleles flag) and the variants in the major histocompatibility complex region were removed. GWAS summary statistics datasets have the sample size column, making the --N flag
unnecessary.Configuration Settings
BIGA offers the option to run LDSC genetic correlation in European GWAS or East Asian GWAS. The LD scores ( used by--w-ld-chr and --ref-ld-chr flags) were from the 1000 Genomes data and were downloaded from LDSC. Specifically:
For European GWAS: Use
--w-ld-chr eur_w_ld_chr/ --ref-ld-chr eur_w_ld_chr/For East Asian GWAS: Use
--w-ld-chr eas_ldscores/ --ref-ld-chr eas_ldscores/Default Script
The script is for running and summarizing the results of LD Score Regression (LDSC).
python2 $ldsc_path/ldsc.py --ref-ld-chr $ref_chr --w-ld-chr $w_chr --out $ldsc_path/log/$job_id/one --rg $summary_statistics1,$summary_statistics2'ukb_phase1to3_ica_node_may_2022_imputed_pheno10_harmo.sumstats.gz',$summary_statistics2'ukb_phase1to3_ica_node_may_2022_imputed_pheno41_harmo.sumstats.gz',$summary_statistics2'ukb_phase1to3_ica_node_may_2022_imputed_pheno29_harmo.sumstats.gz',...
cd $ldsc_path/log/$job_id/
mkdir less
for file in *log
do
sed -n -e '/Summary of Genetic Correlation Results/,/Analysis finished/ p' ${file} > less/less_${file}
awk 'NR>2 && NR<79{print}' less/less_${file} > less/temp.txt ; mv less/temp.txt less/less_${file}
done
The first part involves LDSC execution.
ref-ld-chr $ref_chr and --w-ld-chr $w_chr
specify the reference LD scores and their weights, which vary by the population. --out
$ldsc_path/log/$job_id/one determines the output directory and filename prefix for the LDSC results
for each job. --rg is used to instruct LDSC to calculate genetic correlation. $summary_statistics1
is the path to the input file. $summary_statistics2 is the path prefix of the curated dataset.
Each file in this dataset represents different phenotypes, and LDSC will calculate the genetic correlations
between the input file and the curated datasets.
The second part summarizes the LDSC results.
sed -n -e '/Summary of Genetic Correlation
Results/,/Analysis finished/ p' ${file}: This command extracts a specific section from each log file,
starting from where it finds the phrase "Summary of Genetic Correlation Results" and ending at "Analysis
finished". This section is then redirected to a new file in the less directory. awk 'NR>2 &&
NR<79{print}' less/less_${file}: This awk command further processes each extracted section, selecting
only specific lines (from line 3 to 78), which presumably contain the relevant summarized data. The result
is temporarily saved in less/temp.txt. Afterwards, BIGA will save all the results into the database.
3.2 Massive SumHer Analysis
Reference
SumHer: ldak5.2Curated Dataset
BIGA offers 7 European datasets and 2 East Asian datasets for SumHer genetic correlation analysis. These datasets include:1. Brain and Organ Imaging Traits
2. PGC and and Other Brain Disorders
3. FinnGen
4. UK Biobank (Neale Lab)
5. Biobank Japan
6. UKB-Oxford Brain Imaging Traits
7. GWAS Catalog
8. UKB-PPP European Protein GWAS
9. CHIMGEN
Input File Build
The standard input file format for SumHer is a text file with a header row. BIGA's unified format meets SumHer's requirements for the input file. Columns such aschr (chromosome),
position, a1 (allele 1), a2 (allele 2), n (sample size),
and statistical measures like beta or odds_ratio and se (standard
error) are extracted from this universal format. These are then transformed into a format suitable for
SumHer: chr:position is labeled as Predictor, a1 as A1,
a2 as A2, and n remains. The statistical measures are converted into
Z statistics, either using beta/se or log(odds_ratio)/se. Additionally, the snp
column filters out SNPs that are not part of the HapMap3 dataset (approximately 1.2 million SNPs). After preparing
these columns, BIGA removes any duplicate Predictors to retain only the first occurrence and keeps rows
with single-character alleles (A, C, G, T).Configuration Settings
1. Reference Taggings File: SumHer requires a tagging file for estimating genetic correlations. BIGA uses the pre-computed LDAK-Thin Tagging File as recommended in SumHer including LDAK-Thin Tagging File (GBR population, HapMap3 SNPs), LDAK-Thin Tagging File (EAS population, HapMap3 SNPs) for two populations - the GBR (European) and EAS (East Asian) populations.2. Phenotypic Variance Cutoff: BIGA adheres to SumHer's recommendation and applies a cutoff of 0.01. This means that it removes any predictors that account for more than 1% of the phenotypic variance.
3. Ambiguous Alleles: Since the data is harmonized, ambiguous alleles (A&T or C&G) were aligned with the latest dbsnp 156 reference file. It is not necessary to remove ambiguous alleles, BIGA allows these alleles with the setting
--allow-ambiguous
YES. 4. SNP Availability: To address cases where some SNPs in the summary statistics might not be present in the tagging file, BIGA uses the setting
--check-sums NO to avoid related errors.Default Script
The provided script automates the process of genetic correlation analysis using SumHer, working with various datasets:
(arr=("finngen_R9_O15_PREG_OTHER_MAT_DISORD_GRCh37_CrossMap_harmo.txt",...,"finngen_R9_PAIN_GRCh37_CrossMap_harmo.txt")
for trait2 in "${arr[@]}"; do
out=$sumHer_path/log/$job_id/$trait2
$sumHer_path/ldak5.2.$envir --summary $summary_statistics1 --summary2 $summary_statistics2/$trait2 --sum-cors $out --tagfile $sumHer_path/ldak.thin.hapmap.$popu.tagging --check-sums NO --allow-ambiguous YES
done) &
(arr=("finngen_R9_M13_ARTHROSIS_KNEE_GRCh37_CrossMap_harmo.txt" ,...,"finngen_R9_E4_LIPOPROT_GRCh37_CrossMap_harmo.txt")
for trait2 in "${arr[@]}"; do
out=$sumHer_path/log/$job_id/$trait2
$sumHer_path/ldak5.2.$envir --summary $summary_statistics1 --summary2 $summary_statistics2/$trait2 --sum-cors $out --tagfile $sumHer_path/ldak.thin.hapmap.$popu.tagging --check-sums NO --allow-ambiguous YES
done) &
wait
$summary_statistics1 represents the input summary data, either uploaded or queried by the user.
$summary_statistics2 is the directory where BIGA's curated data is stored. $trait2
refers to specific trait filenames from the FinnGen dataset in this case. $popu is a variable
indicating the population-specific GWAS dataset, which can be either 'gbr' for the European population or
'eas' for East Asian. The script employs a parallel processing approach, indicated by & at the
end of each command. Two arrays of filenames (`arr`) are iterated over, and for each file, a SumHer analysis
is conducted using the ldak5.2.$envir command. $envir can either be
.mac or .linux depending on the operation system. This command processes both the
user's input data and BIGA's curated data, applying the necessary configuration settings, like tagging file
selection (--tagfile), allowing ambiguous alleles (--allow-ambiguous YES), and
bypassing SNP checks (--check-sums NO). The wait command at the end ensures that
all parallel processes are completed before moving on.
3.3 Massive LAVA Analysis
Reference
LAVA: version 0.1.0Curated Dataset
BIGA offers 7 European datasets for LAVA local genetic correlation analysis. These datasets include:1. Brain and Organ Imaging Traits
2. PGC and and Other Brain Disorders
3. FinnGen
4. UK Biobank (Neale Lab)
5. UKB-Oxford Brain Imaging Traits
6. GWAS Catalog
7. UKB-PPP European Protein GWAS
In each curated dataset, users currently have the option to select up to 10 traits for running LAVA’s massive analysis.
Input File Build
The standard input file format for LAVA is a text file with a header row. BIGA's unified format meets LAVA's requirements for the input file. Columns includingsnp, a1,a2,n,beta
or odds_ratio,pvalue are extracted from this universal format. These are then
transformed into a format suitable for LAVA: snp is labeled as SNP,
a1 as A1 and a2 and A2, n as N,
beta as BETA, pvalue as P, odds_ratio as
OR. Furthermore, the SNPs in the input file are filtered using reference data from Phase 3 of
the 1000 Genomes Project. LAVA loci can be defined using genomic coordinates or a list of SNPs, and BIGA
uses the same loci definition as LAVA, utilizing the 1000 Genomes Project data (Phase 3, build
GRCh37/hg19). Sample overlap file is set to NULL.
Default Script
testPath = trait1_path
testName = t1_name
infoFile = paste('data/',job_id,'/','input.info.txt',sep = "")
sink(infoFile)
cat("phenotype cases controls filename\n")
temp = paste(testName,"NA","NA",testPath,'\n',sep=" ")
cat(temp,append = FALSE)
sink(NULL)
trait1 = process.input(input.info.file=infoFile,
sample.overlap.file=NULL,
ref.prefix="vignettes/data/ldsc_g1000_eur",phenos=c(t1_name))
trait1_path and t1_name). The infoFile
is a configuration file that details the input file, including cases and controls set to NA, along with
their respective filenames. The process.input function is then called to process this input
data, considering the reference file from phase 3 of the 1000 Genomes Project (ref.prefix).
if (n.run>0){
for (i in 1:n.run) {
start_time <- Sys.time()
# run all lava for each run
for (j in 1:batch_size) {
if (j == 1){
input = readRDS(paste(lava_summary,'/',trait2_names[(i-1)*batch_size+j],sep = ""))
input = appendEnv(trait1,input)
}else{
tmp = readRDS(paste(lava_summary,'/',trait2_names[(i-1)*batch_size+j],sep = ""))
input = appendEnv(input,tmp)
}
}
data_name = paste0((i-1)*batch_size+1,'_',i*batch_size,sep="")
# Read in locus info file
loci = read.loci("support_data/blocks_s2500_m25_f1_w200.GRCh37_hg19.locfile")
n.loc = nrow(loci)
### Analyse
progress = ceiling(quantile(1:n.loc, seq(.05,1,.05)))
b=u=list()
for (i in 1:n.loc) {
if (i %in% progress)print(paste("..",names(progress[which(progress==i)])))
try({locus = process.locus(loci[i,], input)})
if(!is.null(locus)){
# extract some general locus info for the output
loc.info = data.frame(locus = locus$id, chr = locus$chr, start = locus$start, stop = locus$stop, n.snps = locus$n.snps, n.pcs = locus$K)
if (t1_name %in% locus$phenos){
if(length(locus$phenos)>1) {
# run and bivariate tests
out = run.bivar(locus, target=t1_name)
if(!is.null(out$bivar)) {
b[[i]] = cbind(loc.info, out$bivar)}}}}}}}
n.run), each corresponding to a different trait. For each trait, the script
reads loci information and processes each locus. The run.bivar function performs bivariate
analysis on each locus, comparing it against the target phenotype (t1_name). The results are
accumulated in the variable b, which holds the analysis outcomes.
dataframes = [pd.read_csv(file) for file in file_paths]
df = pd.concat(dataframes, ignore_index=True)
file_paths) into Pandas DataFrames and then
concatenates them into a single DataFrame (df). Each file is expected to be in CSV format.
dfBand = pd.read_csv(lava_path+'/vignettes/data/name/cytoBand.csv', header=0)
bands1, bands2, bands3 = [], [], []
#df = df.reset_index()
for index, row in df.iterrows():
chrIndex = "chr"+str(row["chr"])
start = int(row["start"])
end = int(row["stop"])
band = dfBand[(dfBand["chr"] == chrIndex) & (dfBand["start"] <= start) & (dfBand["end"] >= end)]
if band.empty:
bands = dfBand[(dfBand["chr"] == chrIndex) & (dfBand["start"] <= end) & (dfBand["end"] >= start)]
names = list(bands["name"])
if bands.empty:
bands1.append("NA")
bands2.append("NA")
bands3.append("NA")
elif len(bands) <= 2:
if (int(list(bands["start"])[1]) - start) >= (end - int(list(bands["start"])[1])):
bands1.append(names[0])
bands2.append(names[1])
bands3.append("NA")
else:
bands1.append(names[1])
bands2.append(names[0])
bands3.append("NA")
else:
if (int(list(bands["start"])[1]) - start) >= (end - int(list(bands["start"])[2])):
bands1.append(names[1])
bands2.append(names[0])
bands3.append(names[2])
else:
bands1.append(bands["name"][1])
bands2.append(bands["name"][2])
bands3.append(bands["name"][0])
else:
name = list(band["name"])
bands1.append(name[0])
bands2.append("NA")
bands3.append("NA")
df["bands1"] = bands1
df["bands2"] = bands2
df["bands3"] = bands3
cytoBand.csv) and iterates through each row of the main DataFrame
(df). For each row, it determines the chromosomal bands (bands1,
bands2, bands3) that overlap with the genomic region specified by
start and stop in the row. This is done by comparing the row's genomic region with
those in the chromosomal band data (dfBand).
The script filters the results for significance based on the p-value. It drops any rows with missing data and keeps only those rows with p-value less than or equal to 0.05, indicating statistical significance. These filtered results are then saved to another CSV file (
significant.csv),
which contains only significant results. These results will be displayed on the BIGA result table page.
3.4 Massive Popcorn Analysis
Reference
Popcorn: version 1.0.0 (python3.7)Curated Dataset
BIGA offers the Biobank Japan and CHIMGEN datasets for Popcorn cross-ancestry genetic correlation analysis. Therefore, if you want to run a Popcorn analysis, you will need to upload European summary statistics data to conduct cross-ancestry genetic correlation analysis.Input File Build
The standard input file format for Popcorn is a text file with a header row. BIGA's unified format meets Popcorn's requirements for the input file. Columns includingsnp,
a1,a2,n,beta, se,
odds_ratio,pvalue,eaf are extracted from this universal format. These
are then transformed into a format suitable for Popcorn: snp is labeled as SNP,
a1 and a2 remains, n as N, beta remains,
se as SE, pvalue as p-value, odds_ratio as
OR. Additionally, Popcorn only requires that the effect allele to be the same column in the file
for both populations (e.g. a1 as effect in both input files from pop1 and pop2). The parameters
AF in Popcorn can be calculated by 1-eaf since AF is the allele
frequency of a2 and eaf is the frequency of a1
Configuration Settings
BIGA uses pre-calculated cross-population scores provided by Popcorn (https://www.dropbox.com/sh/37n7drt7q4sjrzn/AAAa1HFeeRAE5M3YWG9Ac2Bta). The score is for EUR and EAS 1000 genomes populations. When computing heritability and trans-ethnic genetic correlation for two sets of summary statistics data, BIGA uses the function ofFit mode in Popcorn. The default estimator is
regression, following Popcorn's recommendation due to the relative instability of the maximum likelihood
estimation (MLE) methods. You can select between Regression and MLE for the
estimator. Another consideration in Popcorn is whether to compute the genetic effect correlation or the
genetic impact correlation. BIGA uses --gen_effet as default since Popcorn believes that it's a more
realistic model. BIGA uses MAF threshold of 0.05 by default since the larger the cutoff, the closer the
genetic effect and the genetic impact results will be.
Default Script
The provided script automates the process of genetic correlation analysis using Popcorn, working with Biobank Japan datasets:
arr=("GWASsummary_BrC_Japanese_SakaueKanai2020.auto_harmo.txt",...,"GWASsummary_SAS_Japanese_SakaueKanai2020.auto_harmo.txt")
if [ $use_mle == "yes" -a $use_gen_effect == "yes" ]; then
for trait2 in "${arr[@]}"; do
out=$popcorn_path/log/$job_id/$trait2
python -m popcorn fit -v 1 --use_mle --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --gen_effect --sfile1 $summary_statistics1 --sfile2 $biobankjapan_dir/$trait2 $out
done
elif [ $use_mle == "yes" -a $use_gen_effect == "no" ]; then
for trait2 in "${arr[@]}"; do
out=$popcorn_path/log/$job_id/$trait2
python -m popcorn fit -v 1 --use_mle --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --sfile1 $summary_statistics1 --sfile2 $biobankjapan_dir/$trait2 $out
done
elif [ $use_mle == "no" -a $use_gen_effect == "yes" ]; then
for trait2 in "${arr[@]}"; do
out=$popcorn_path/log/$job_id/$trait2
python -m popcorn fit -v 1 --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --gen_effect --sfile1 $summary_statistics1 --sfile2 $biobankjapan_dir/$trait2 $out
done
else
for trait2 in "${arr[@]}"; do
out=$popcorn_path/log/$job_id/$trait2
python -m popcorn fit -v 1 --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --sfile1 $summary_statistics1 --sfile2 $biobankjapan_dir/$trait2 $out
done
fi
$summary_statistics1 represents the input summary data, either uploaded or queried by the user.
$biobankjapan_dir is the directory where the curated data is stored. $trait2
refers to specific trait filenames from the Biobank Japan dataset. EUR_EAS_all_gen_imp.cscore
is the pre-calculated score. The script executes a series of conditional operations based on user-specified
parameters (use_mle, use_gen_effect). It iterates through an array of genetic
traits (listed in arr), performing the specified analysis for each trait. The if,
elif, and else statements direct the flow of execution.
3.5 Pairwise Analysis
1. Input File Build
Pairwise analysis allows you to upload/query two GWAS summary statistics data. For each analysis, you can choose one method from a selection that includes LDSC, SumHer, LAVA, and Popcorn. The process involves harmonizing the input files and converting them into the appropriate format for the selected analysis method. The input files will do harmonization steps and transition to the corresponding format to run different methods. The input file build process for each method is the same as in the corresponding Massive Analysis section (LDSC, SumHer, LAVA, Popcorn)2. LDSC
Parameter settings: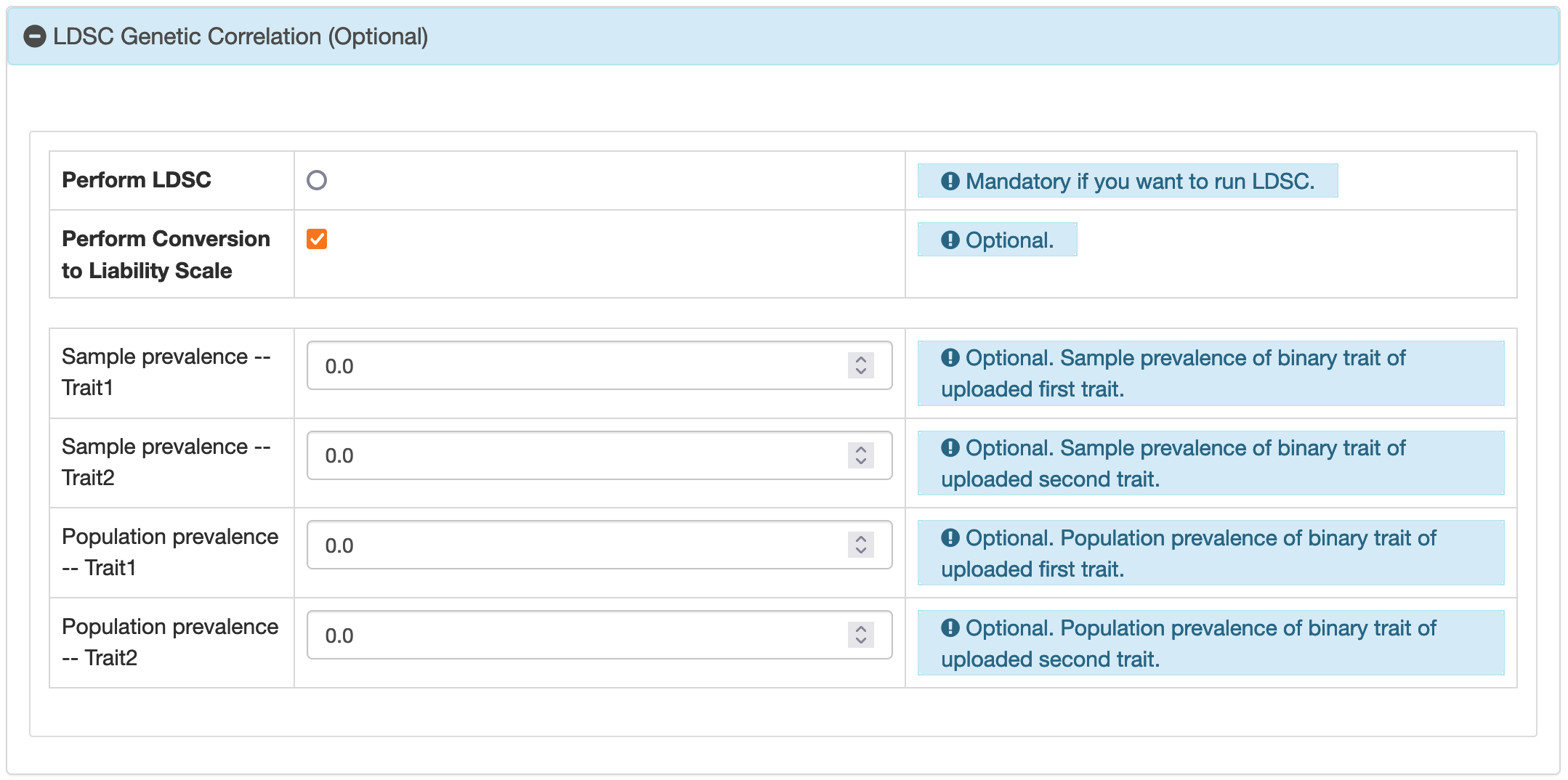
Default Script:
The provided script automates the process of genetic correlation analysis between two traits using LDSC:
if [ $samp_prev == "None" ]; then
python2 $ldsc_path/ldsc.py --rg $summary_statistics1,$summary_statistics2 --ref-ld-chr $ref_chr --w-ld-chr $w_chr --out $out
else
python2 $ldsc_path/ldsc.py --rg $summary_statistics1,$summary_statistics2 --ref-ld-chr $ref_chr --w-ld-chr $w_chr --out $out --samp-prev $samp_prev --pop-prev $pop_prev
fi
Here, $summary_statistics1 and $summary_statistics2 represent the file paths for
the first and second traits, respectively. When you opt for conversion to the Liability scale, additional
flags --samp-prev and --pop-prev are included in the command to incorporate the
prevalence parameters.
3. SumHer
Parameter settings: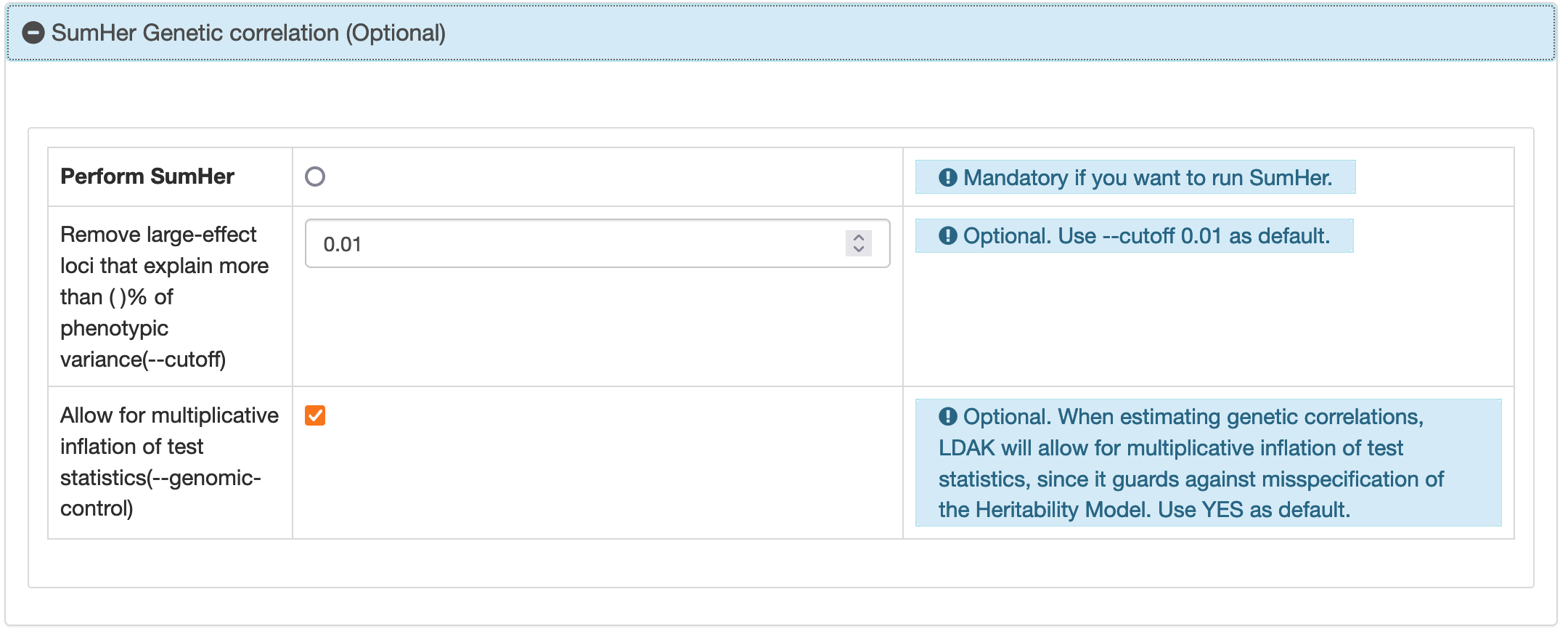
Default Script:
The provided script automates the process of genetic correlation analysis between two traits using SumHer:
$sumHer_path/ldak5.2.$envir --summary $summary_statistics1 --summary2 $summary_statistics2 --sum-cors $out --tagfile $sumHer_path/ldak.thin.hapmap.$popu.tagging --check-sums NO --allow-ambiguous YES --cutoff $cutoff --genomic-control $geno_control
$summary_statistics1 represents the first input summary data, either uploaded or queried by the
user. $summary_statistics2 represents the second input summary data. $popu is a
variable indicating the population-specific GWAS dataset, which can be either 'gbr' for the European
population or 'eas' for East Asian. A SumHer analysis is conducted using the ldak5.2.$envir
command. $envir can either be .mac or .linux depending on the
operating system. The parameter $cutoff is employed to determine the threshold percentage for
excluding loci with significant effects. Finally, the option --genomic-control is set to either
YES or NO to decide on the allowance for multiplicative inflation in the test statistics.
4. LAVA
Parameter settings: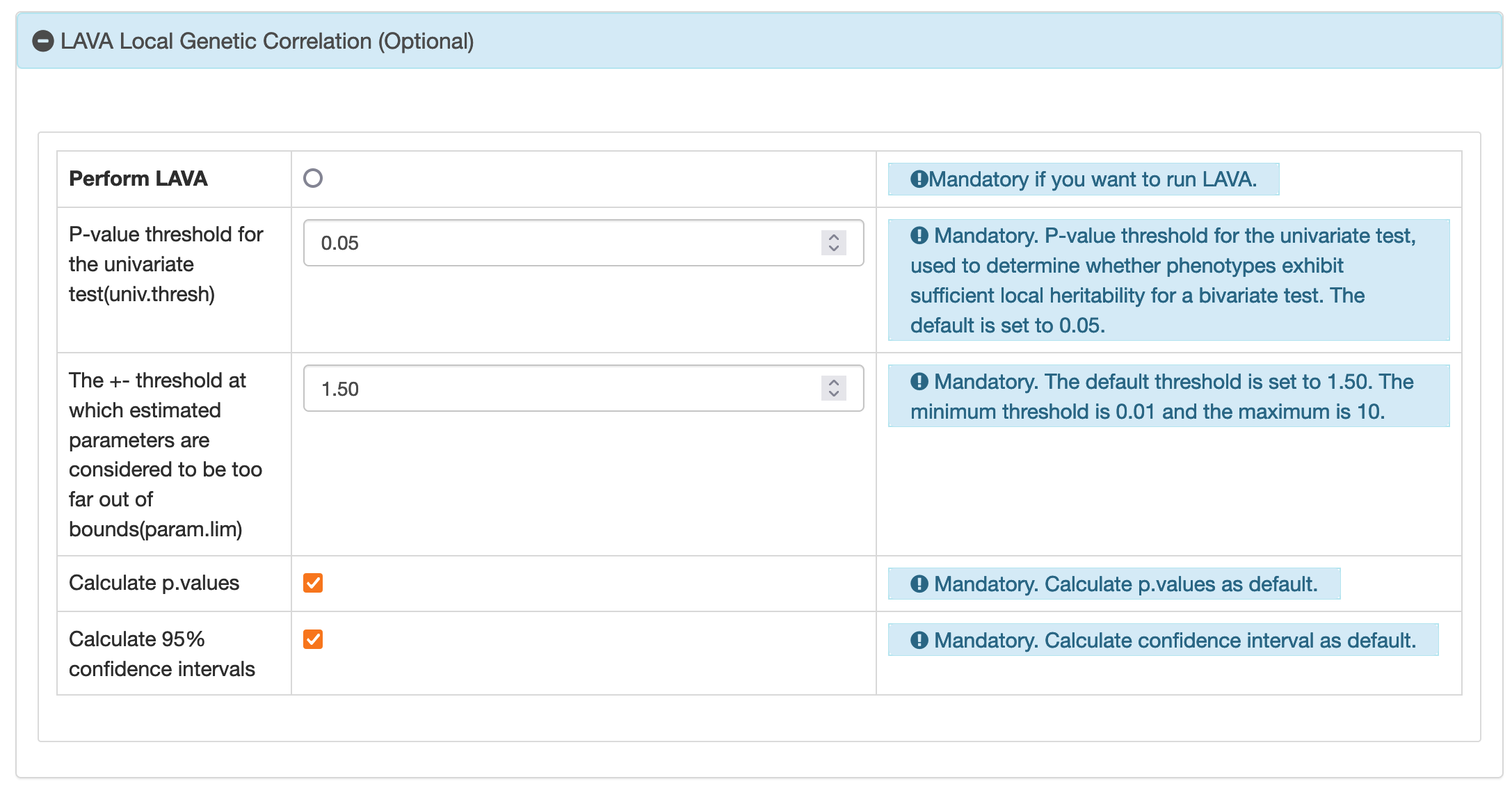
run.univ.bivar(locus, univ.thresh=uinv_thresh,
p.values=p_values, CIs=ci, param.lim=parlim)
Default Script:
Rscript $lava_path/shell/2traits.R $lava_path $summary_statistics1 $summary_statistics2 $job_id $t1_name $t2_name $univ_thresh $p_values $ci $parlim --no-restore --no-save
$summary_statistics1 represents the first input summary data, either uploaded or
queried by the user. $summary_statistics2 represents the second input summary data. job_id
is your job running id. t1_name,t2_name are the traits' names. univ_thresh is the
p-value threshold for the univariate test. If ci is set to 'YES', then calculate confidence
intervals. If p_values is set to `YES`, then calculate p-values. parlim is the +-
threshold for estimated parameters.
5. Popcorn
Parameter settings: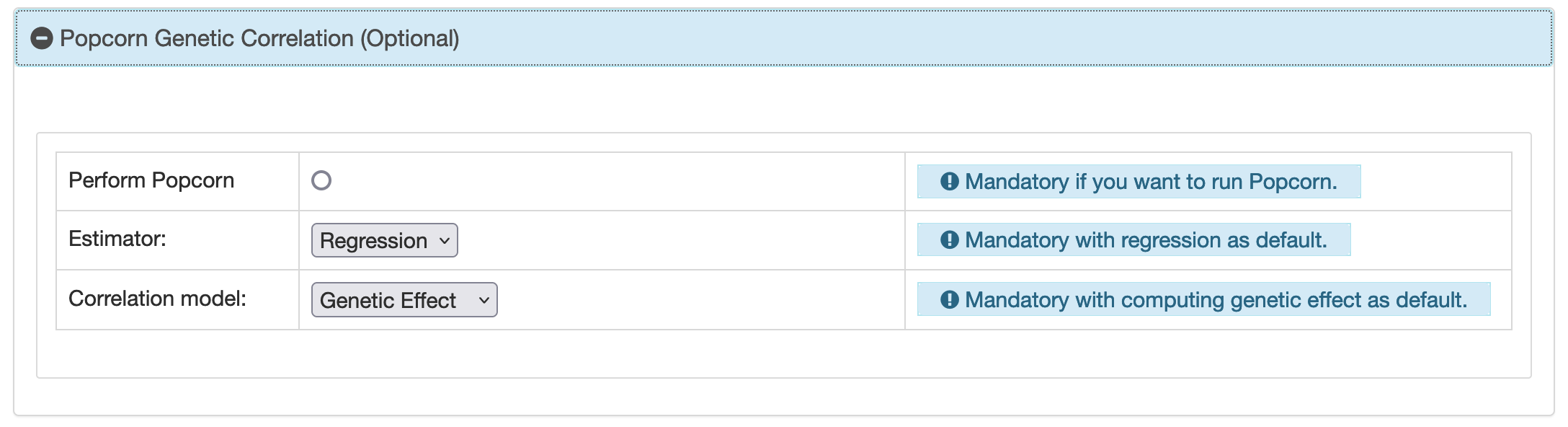
Default Script:
if [ $use_mle == "use_mle" -a $use_gen_effect == "use_gen_effect" ]; then
out=$popcorn_path/2traits/log/$job_id/mle_gen_effect
python -m popcorn fit -v 1 --use_mle --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --gen_effect --sfile1 $summary_statistics1 --sfile2 $summary_statistics2 $out
elif [ $use_mle == "use_mle" -a $use_gen_effect == "use_gen_impact" ]; then
out=$popcorn_path/2traits/log/$job_id/mle_gen_imp
python -m popcorn fit -v 1 --use_mle --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --sfile1 $summary_statistics1 --sfile2 $summary_statistics2 $out
elif [ $use_mle == "use_regression" -a $use_gen_effect == "use_gen_effect" ]; then
out=$popcorn_path/2traits/log/$job_id/mle_reg_effect
python -m popcorn fit -v 1 --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --gen_effect --sfile1 $summary_statistics1 --sfile2 $summary_statistics2 $out
else
out=$popcorn_path/2traits/log/$job_id/mle_reg_imp
python -m popcorn fit -v 1 --cfile $popcorn_path/ref/EUR_EAS_all_gen_imp.cscore --sfile1 $summary_statistics1 --sfile2 $summary_statistics2 $out
fi
$summary_statistics1 represents the first input summary data, either uploaded or queried by the
user. $summary_statistics2 represents the second input summary data. EUR_EAS_all_gen_imp.cscore
is the pre-calculated score. The script executes a series of conditional operations based on user-defined
parameters (use_mle, use_gen_effect). The `if`, `elif`, and `else` statements
direct the flow of execution.
4. Analysis Results and Log Files
4.1 Analysis Results
The pairwise analysis result page shows similar information. If your job is successfully completed, you can download the entire set of results by clicking the "download" link, marked as "result$job_id.tar.gz". This package also contains the BIGA log file, the method run log file, and the output of the method you used. In instances where your job fails, the log file is accessible for review to determine the cause of the failure.
4.2 Read BIGA Log Files
ieu-b-30 from IEU OpenGWAS and then running Massive
LDSC Analysis with PGC dataset.
************************************************************************
* BigaGWAS WorkShop
* version 1.1.0
* Yujue Li and Bingxin Zhao (2023-2024)
* Purdue University and University of Pennsylvania
* GNU General Public License v3
************************************************************************
This section is the masthead. It contains basic information about the software, including its name, version,
authors, the associated institutions, and the license under which it is distributed.
************************************************************************
******************* Task Type: Massive LDSC Analysis *******************
************************************************************************
Massive Analysis: Method=massive_ldsc Job_type=group_pgc Job_id=995.
************************************************************************
******************** Task ID and Job Configurations ********************
************************************************************************
Task running ID: 9091bd78-1232-4391-9fed-24c5cf66c694.
Current Job Settings:{'title': 'GCST90000060', 'username': 'admin', 'trait1_name': 'GCST90000060', 'job_id': '995', 'email_to': 'li4476@purdue.edu', 'need_harmonize': False, 'dropna': True, 'header': None, 'snp': False, 'query_error': False, 'chunksize_map_rsid': 200000, 'chunksize_harmo': 200000, 'gwas_catalog_chunksize': 10000, 'chunksize': 100000, 'convert_back_to_hg19': True, 'data_dir': '/Users/yujue/Documents/githubPackage/ldsc/data/995', 'log_dir': '/Users/yujue/Documents/githubPackage/ldsc/log/995', 'summary1_path': '/Users/yujue/Documents/githubPackage/ldsc/data/995/995_ldsc.sumstats.gz', 'compression': 'txt', 'job_type': 'massive_ldsc', 'choose_type': 'group_pgc', 'value': 'pgc', 'LDSC_PATH': '/Users/yujue/Documents/githubPackage/ldsc', 'population': 'european', 'group': 'PGC_and_Brain_Disorders', 'add_prev': False, 'samp_prev': 'nan', 'pop_prev': 'nan', 'chr': None, 'position': None, 'rsid': None, 'n': None, 'a1': None, 'a2': None, 'beta': None, 'eaf': None, 'pvalue': None, 'se': None, 'odds_ratio': None, 'zscore': None, 'hg_version': None, 'hg19_final_data': '/Users/yujue/Documents/githubPackage/ldsc/data/995/995.txt', 'hg19_final_data_gz': '/Users/yujue/Documents/githubPackage/ldsc/data/995/995.txt.gz', 'link1': 'http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics', 'sample_size_catalog': '55134', 'query_party': 'gwas_catalog', 'select_trait1': 'querying', 'query_id': 'GCST90000060', 'celery_id': '9091bd78-1232-4391-9fed-24c5cf66c694'}.
In this section, the log file provides an overview of the current analysis being run. It includes the method
used (`massive_ldsc`), the type of job (`group_pgc`), and a unique job ID (`758`). There's also a task
running ID, which is likely a unique identifier for this specific instance of the task.
************************************************************************
********************** Querying from IEU OpenGWAS **********************
************************************************************************
Task running script:
sh /Users/yujue/Documents/githubPackage/bigagwas/static/shell/query.sh ebi-a-GCST007517 /Users/yujue/Documents/githubPackage/ldsc/data/988 https://gwas.mrcieu.ac.uk/files /opt/homebrew/anaconda3/etc/profile.d/conda.sh ldsc /Users/yujue/Documents/githubPackage/query 298957
ebi-a-GCST007517
/Users/yujue/Documents/githubPackage/ldsc/data/988
https://gwas.mrcieu.ac.uk/files
/opt/homebrew/anaconda3/etc/profile.d/conda.sh
ldsc
/Users/yujue/Documents/githubPackage/query
298957
Read VCF File.
Finish reading VCF file.
Starting to convert into text file for harmonization.
OpenGWAS file process complete. Running time: 1.3004319667816162
....
Queried Data Download Finished. Time consuming: 9.6406888961792 seconds.
************************************************************************
******************* Checking Input data from OpenGWAS ******************
************************************************************************
Queried data includes columns: ['CHR', 'POS', 'SNPID', 'A2', 'A1', 'ebi-a-GCST007517', 'BETA', 'SE', 'LP', 'AF1', 'P_value', 'N']
Input data check Finished.
This part details the querying process from IEU OpenGWAS, including script execution and file processing. It
provides information on reading the downloaded VCF files and the time taken to complete these processes.
Here, it is focused on converting data into the format for harmonization.
*********** Starting to map rsid to chromosome and position ************
Mapping dbSNP156_GRCh38_chr_12-3.txt : 443.08141112327576 seconds
Mapping dbSNP156_GRCh38_chr_10-3.txt : 480.74914479255676 seconds
Mapping dbSNP156_GRCh38_chr_14-1.txt : 582.9732220172882 seconds
...
************************************************************************
********************* Harmonization: Mapping rsid *********************
************************************************************************
Mapping dbSNP156_GRCh38_chr_10-2.txt : 23.715790033340454 seconds
Mapping dbSNP156_GRCh38_chr_10-1.txt : 26.09787106513977 seconds
Mapping dbSNP156_GRCh38_chr_14-2.txt : 27.18862295150757 seconds
Mapping dbSNP156_GRCh38_chr_14-1.txt : 28.28748393058777 seconds
...
Overall Forward strand rate: 1.0, Reverse strand rate:0.0.
************************************************************************
************ Harmonization: Harmonizing data by chromosome *************
************************************************************************
Chromosome 5, Strand Inference Summary:
Counter({'Forward strand variant': 5421, 'Palindormic variant': 696})
Chromosome 5, Harmonization summary:
Counter({'Forward strand; Correct orientation; Already harmonised': 5419, 'Palindromic; Assume forward strand; Correct orientation; Already harmonised': 695, 'Forward strand; Flipped orientation; Requires harmonisation': 2, 'Palindromic; Assume forward strand; Flipped orientation; Requires harmonisation': 1})
Chromosome 7, Strand Inference Summary:
...
Harmonization ends. Time consuming: 182.41979694366455 seconds.
This section covers the mapping of rsid to chromosome and position aligned with GRCh38 and the time taken
for each chromosome. It then goes into harmonizing the summary statistics data, inferring the direction of
the strand, providing detailed summaries and counts for each chromosome. The section ends by noting the
total time spent on harmonization.
************************************************************************
*************** Combining and Sorting Harmonized data ******************
************************************************************************
Combining and Sorting ends. Time consuming: 0.9061808586120605 seconds.
This section indicates that the harmonized data has been combined and sorted by chromosomes. The time taken
for this process is noted, allowing for performance tracking.
************************************************************************
************ Starting to convert back to GRCh37 version ****************
************************************************************************
0 positions did not align with the GRCh37 reference.
Liftover to GRCh37 Finished. Time Consuming: 4.547966957092285 seconds.
Here, the log file records the process of converting the data back to the GRCh37 for analysis because LDSC
needs position built on GRCh37 version. It specifically notes that 1366 positions did not align with the
GRCh37 reference file. The duration of this liftover process is also recorded.
************************************************************************
********* Converting into sumstats.gz for use in LDSC analysis *********
************************************************************************
/Users/yujue/Documents/githubPackage/ldsc/data/988/988.txt.gz
/Users/yujue/Documents/githubPackage/ldsc/data/988/988_ldsc
/opt/homebrew/anaconda3/etc/profile.d/conda.sh
py27
/Users/yujue/Documents/githubPackage/ldsc
no
*********************************************************************
* LD Score Regression (LDSC)
* Version 1.0.1
* (C) 2014-2019 Brendan Bulik-Sullivan and Hilary Finucane
* Broad Institute of MIT and Harvard / MIT Department of Mathematics
* GNU General Public License v3
*********************************************************************
This part of the log indicates the final conversion of the data into `sumstats.gz` format, which is used in
LDSC analysis. The time taken for this conversion is provided.
************************************************************************
************************* Running LDSC Software ************************
************************************************************************
Starting to run 24 PGC psychiatric disorders traits.
Command line:
sh /Users/yujue/Documents/githubPackage/bigagwas/static/shell/pgc_24.sh 988 /Users/yujue/Documents/githubPackage/ldsc/data/988/988_ldsc.sumstats.gz /Users/yujue/Documents/githubPackage/ldsc /Volumes/sandisk/summary_statistics/harmo/ldsc/data/pgc/hg19/ /opt/homebrew/anaconda3/etc/profile.d/conda.sh py27 /eur_w_ld_chr/
This section shows the initiation of the LDSC software to analyze 24 PGC psychiatric disorders and other brain diseases traits. It
includes the command line used to run the analysis.
*********************************************************************
* LD Score Regression (LDSC)
* version 1.1.0
* (C) 2014-2019 Brendan Bulik-Sullivan and Hilary Finucane
* Broad Institute of MIT and Harvard / MIT Department of Mathematics
* GNU General Public License v3
*********************************************************************
Call:
./ldsc.py \
--ref-ld-chr /Users/yujue/Documents/githubPackage/ldsc/eur_w_ld_chr/ \
--out /Users/yujue/Documents/githubPackage/ldsc/log/758/phenoone \
--rg /Users/yujue/Documents/githubPackage/ldsc/data/758/758_ldsc.sumstats.gz,/Volumes/sandisk/summary_statistics/harmo/ldsc/data/pgc/hg19/8_ocd_aug2017.sumstats.gz
...
Finally, the log file gives the output of the LDSC software being used. More information about this section is
provided by LDSC software itself.
5. Queue System and Periodic Jobs
5.1 Queue System
5.2 Periodic Jobs
Daily GWAS Catalog Update
This function is scheduled every day at 5:30 AM Eastern Time to keep the GWAS Catalog available list up-to-date. The GWAS Catalog offers a daily updated harmonized data list (http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics), and in order to maintain synchronization with the GWAS Catalog, BIGA will also refresh its list on a daily basis.Daily Input Files Cleanup
This function is scheduled every day at 5:30 AM Eastern Time to clean out input files stored in BIGA for more than 7 days. Considering that the summary statistics data typically exceeds 500 MB, this process is crucial for maintaining efficient data usage, especially with numerous jobs in progress. BIGA temporarily saves your input files for 7 days, after which all input files are deleted based on the last update date of your job. This regular cleanup routine can keep the system running smoothly and ensure the data storage is effectively managed for both current and future users.6. Other Functions
6.1 Email Notification
Please ensure to check your junk or spam email folder as well, in case any notifications were mistakenly directed there.
6.2 Public Data
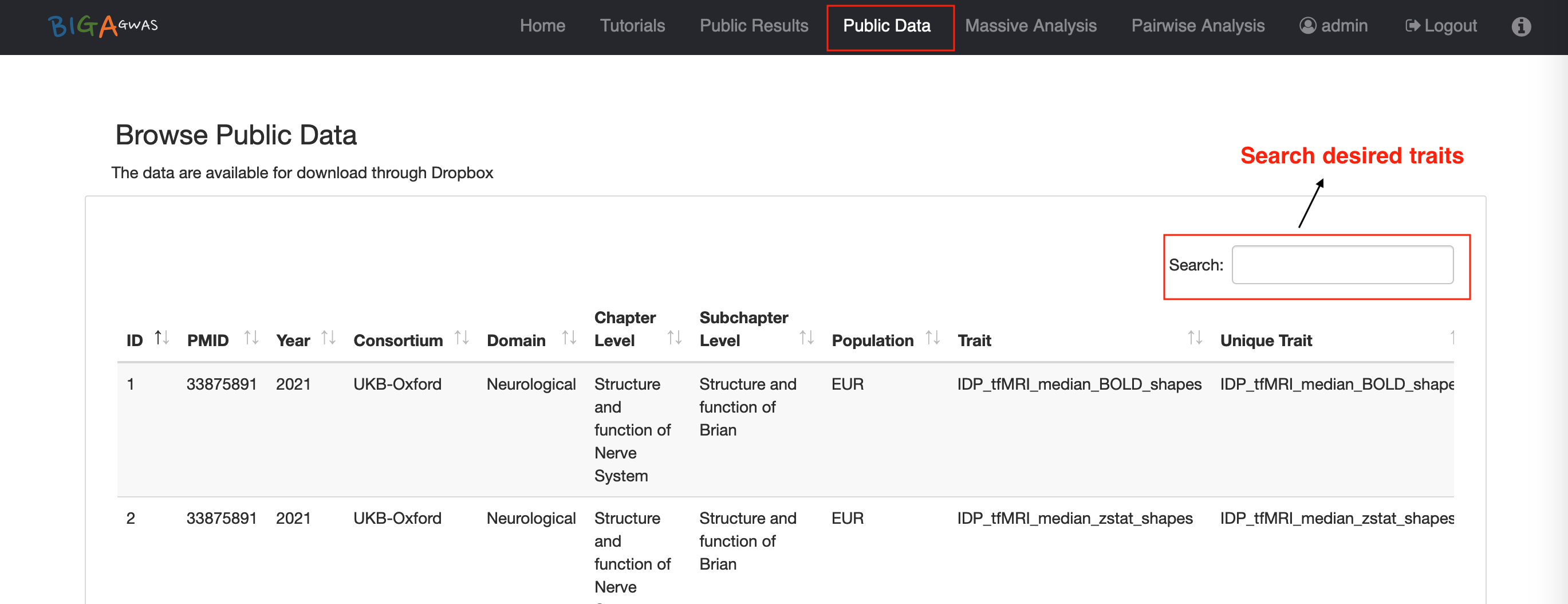
6.3 Public Results
When making results or data publicly available on BIGA, users should be mindful of the risk of data misuse and take appropriate measures to protect participant privacy. Any sensitive information or data that could potentially lead to the re-identification of participants must be excluded from public releases. We strongly recommend that users follow the NIH’s Management of Genomic Summary Results Access Policy (https://grants.nih.gov/grants/guide/notice-files/NOT-OD-19-023.html). Only non-sensitive datasets and summary statistics that meet the NIH’s criteria for public release should be shared through the BIGA platform.